آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Chaperonin |


|
|
Read More
Date: 29-11-2015
Date: 9-12-2020
Date: 6-4-2021
|
Chaperonin
The chaperonins are a family of molecular chaperone found in all types of cell whose function is to assist the correct folding of certain polypeptide chains that have been either newly synthesized or generated from native proteins by environmental stresses that cause partial unfolding.
1. Nomenclature
The term chaperonin was suggested by Sean Hemmingsen (1) to describe a family of highly sequence-related molecular chaperones found in chloroplasts, mitochondria, and eubacteria such as Escherichia coli. This term was proposed to simplify the existing complex nomenclature for different members of this family whose close relationship was only realized when cDNA for the chloroplast chaperonin was sequenced (1). The term was subsequently extended to distant homologues found in archaebacteria and the eukaryotic cytosol (2, 3). The two subfamilies of the chaperonin family are referred to as follows:
1.The GroE or group I subfamily, found in chloroplasts and other plastids, mitochondria, and all eubacteria
2. The TCP-1 or group II subfamily, found in archaebacteria and the eukaryotic cytosol
The GroE terminology reflects the fact that the eubacterial protein was first identified by genetic studies in four laboratories in 1972/73 as a bacterial protein required for the replication of bacteriophage such as lambda in E. coli (4). “Gro” refers to phage growth, and the suffix “E” refers to the observation that the phage growth defect is overcome when the phage carries a mutation in the head gene E. The TCP-1 terminology is derived from the identification of a protein called the t-complex polypeptide encoded by the mouse T locus (5). The mitochondrial members of the GroE subfamily are sometimes referred to as “hsp60” proteins, but this terminology should not be used to describe the chaperonins as a whole, since the eukaryotic TCP-1 members and the chloroplast GroE members are not heat shock proteins.
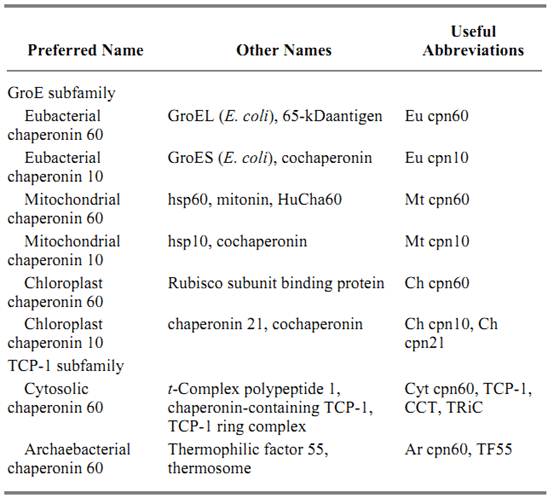
Table 1 presents a suggested nomenclature and useful abbreviations for members in both subfamilies, and lists other names that are used. Some authors confine the abbreviation “cpn60” to the GroE subfamily, but this restriction is inconsistent with the fact that the TCP-1 members share with the GroE members subunit molecular masses of approximately 60 kDa.
Table 1. Nomenclature of the Chaperonins
2. Function
The main function of the chaperonins is to assist the folding of certain newly synthesized polypeptide chains into their biologically active conformations. They achieve this end, not by providing steric information required for each chain to fold correctly, but by sequestering each partially-folded chain in a protected compartment generated by the oligomeric structure of the chaperonin molecule. In this compartment, each chain can continue to fold to a point where aggregation with other partially folded chains is no longer a problem (6-9). This aggregation arises because some proteins fold via intermediate states that expose hydrophobic surfaces transiently and are thus subject to intermolecular interactions with similar states. This aggregation effect can sometimes be observed when chemically denatured proteins refold spontaneously in dilute solution in vitro, but its magnitude is expected to be enhanced in the intact cell because of the phenomenon of macromolecular crowding, or excluded volume, thus increasing the effective concentrations of these states by two or three orders of magnitude (8, 9). It should be stressed that, although the chaperonins increase the yield of correctly folded proteins by minimizing aggregation, they do not increase the rate of folding above that achieved by the fastest folding fraction of spontaneously refolding chains that manage to avoid aggregation.
The best evidence for this view of chaperonin function comes from genetic and pulse-chase labeling studies with living cells. Several proteins imported into the mitochondria of yeast cells form aggregates when the function of the mitochondrial chaperonin is impaired by mutation (10), whereas about 30% of newly synthesized soluble cytoplasmic proteins in E. coli cells become either insoluble or inactive when cpn60 function is switched off by means of a temperature-sensitive mutation (11). In vitro studies of the ameliorating effect of added GroE proteins on the aggregation of various pure denatured proteins during their refolding after removal of denaturant are consistent with this antiaggregation role (12-14). In vitro studies also suggest that a side effect of this antiaggregation role is the partial unfolding of nonaggregated chains that have become kinetically trapped in misfolded conformations, thus allowing these chains another chance to fold correctly (15); it is not clear how important such an effect may be in vivo. In addition, the GroE chaperonins in eubacteria and mitochondria (16) and the TCP-1 chaperonins in the archaebacteria (17) are stress proteins that prevent and reverse the denaturation of some fully folded proteins by stresses such as high temperature.
It must be emphasized that the actual spectrum of proteins that use chaperonin function to fold correctly in intact cells is not known, but preliminary calculations and observations suggest that it is probably a minority in the case of both the GroE chaperonins (9, 18) and the TCP-1 chaperonins (7). Genetic evidence derived from the study of yeast mutants suggests that a major function of the eukaryotic TCP-1 is to assist the folding of tubulin and actin (19), consistent with the observation that newly synthesized chains of actin and tubulin can be isolated from pulse-labeled cells of CHO cells in the form of complexes with TCP-1 chaperonin oligomers (20). In vitro protein refolding experiments also suggest that the substrate specificities of the GroE chaperonins and the TCP-1 chaperonins are different (6).
3. Structure
The chaperonins occur as large oligomeric structures consisting of two stacked rings of subunits, each about 55–60 kDa in size, surrounding a central cavity or cage in which the protein substrate binds; each subunit catalyzes the slow hydrolysis of ATP to ADP. The GroE chaperonins have seven subunits per ring, whereas the TCP-1 chaperonins have eight or nine. The subunit sequence identity between members of the GroE subfamily is in the range 42–76%, whereas that between members of the TCP-1 subfamily is around 30–40%, but the identity between the subfamilies is much less (15-20( % and is confined to regions corresponding to the ATPase domain in GroE cpn60. The evolutionary implications of these similarities are under debate (7, 21) .
A convenient abbreviation for the chaperonin(s) is cpn(s). Thus the subunits of the ring can be called cpn60 subunits, and the oligomer can be called cpn60. The GroE subfamily also contains another type of oligomeric ring made of seven smaller 10-kDa subunits, called cpn10, because there is a slight similarity between the cpn10 subunit sequence and part of the cpn60 subunit sequence (21). GroE cpn60 and cpn10 oligomers bind to each other in 1:1 and 1:2 ratios in the presence of either ATP or ADP, and these binary complexes play an essential role in the protein folding function of these molecules (8, 9). The chloroplast cpn10 oligomer is unusual in that each subunit consists of two copies of a cpn10 sequence fused head to tail and thus is often referred to as cpn21 (22). The TCP-1 oligomers found in the eukaryotic cytosol are much more variable than those found in either the archaebacteria or the GroE subfamily, since subunits of the latter consist of only one or two sequences, whereas the former have at least eight different, but related, subunit sequences in each ring (6, 7, 23). For this reason, the eukaryotic TCP-1 oligomers are also called CCT complexes, for chaperonin-containing TCP-1 complexes, or TriC for TCP-1 ring complexes (see Table 1). The TCP-1 chaperonins do not appear to contain cpn10-like members.
The best-studied chaperonins are the cpn60 and cpn10 oligomers from E. coli, termed GroEL and GroES, respectively. GroEL has been extensively studied by both negative stain and cryoelectron microscopy (24, 25) and by X-ray crystallography of a double mutant form (26). GroEL is a cylindrical structure containing a central cavity about 50 Å in diameter (Fig. 1a). Each subunit consists of (1) an apical domain to which the protein substrate and GroES bind; (2) an equatorial domain that protrudes into the central cavity, which contains the ATPase site and is responsible for most of the intersubunit interactions; and (3) an intermediate domain that contains potential hinge sites responsible for the considerable movements of the other two domains revealed by electron microscopy (24, 25). These conformational changes result from the binding of nucleotide and GroES to the GroEL (26) cylinder. The crystal structure of GroES shows a dome-shaped structure with a hydrophilic inner surface that fits over one end of the GroEL cylinder (24, 27, 28) creating a large enclosed dome-shaped space about 65 × 80 Å in size, inside which the protein substrate continues to fold (29-32) .

Figure 1. Structure and function of the chaperonin system. (a,b) Space-filling representations showing a top and side view, respectively, of the crystal structure of a double mutant form of GroEL (26). Two adjacent subunits are colored with the apical domains in red and purple, the intermediate domains in orange and yellow, and the equatorial domains in blue and green, respectively. Free passage between the GroEL rings is obstructed by N- and C-terminal residues not resolved in the crystal structure. (b) side view of GroES (28) at the top; two adjacent domains and a single mobile loop that is structured in the GroES crystal due to crystal packing are colored. The loop regions normally protrude from the base of GroES downward toward the GroEL. (c) Asymmetrical GroEL/GroES complexes as revealed by cryoelectron microscopy (45). Note the upward and outward movement of the apical GroEL domains interacting with GroES. (d( Model of the GroEL/GroES reaction cycle in assisting protein folding (30); see text for details. The term unfolded protein refers to a partially folded intermediate that is represented by the light pink spheres; folded protein is represented by the dark pink sphere, while the hatched spheres represent a mixture of folded and partially- folded proteins expected in a population of GroEL molecules. At step 4, GroES may associate with either the protein-containing ring of GroEL or with the opposite empty ring; the latter possibility is not shown. Reprinted from Nature 381, 571–580 (1996), with permission; copyright (1996) Macmillan Magazines Ltd.
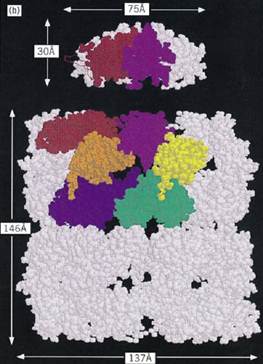
Figure 1. (continued) Structure and function of the chaperonin system. (a,b) Space-filling representations showing a top and side view, respectively, of the crystal structure of a double mutant form of GroEL (26). Two adjacent subunits are colored with the apical domains in red and purple, the intermediate domains in orange and yellow, and the equatorial domains in blue and green, respectively. Free passage between the GroEL rings is obstructed by N- and C-terminal residues not resolved in the crystal structure. (b) side view of GroES (28) at the top; two adjacent domains and a single mobile loop that is structured in the GroES crystal due to crystal packing are colored. The loop regions normally protrude from the base of GroES downward toward the GroEL. (c) Asymmetrical GroEL/GroES complexes as revealed by cryoelectron microscopy (45). Note the upward and outward movement of the apical GroEL domains interacting with GroES. (d) Model of the GroEL/GroES reaction cycle in assisting protein folding (30); see text for details. The term unfolded protein refers to a partially folded intermediate that is represented by the light pink spheres; folded protein is represented by the dark pink sphere, while the hatched spheres represent a mixture of folded and partially- folded proteins expected in a population of GroEL molecules. At step 4, GroES may associate with either the protein-containing ring of GroEL or with the opposite empty ring; the latter possibility is not shown. Reprinted from Nature 381, 571–580 (1996), with permission; copyright (1996) Macmillan Magazines Ltd.
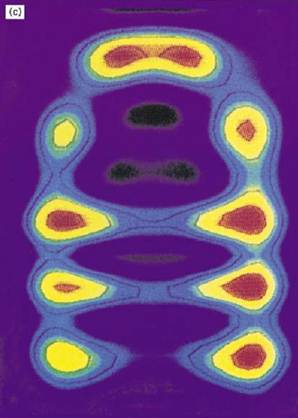
Figure 1. (continued) Structure and function of the chaperonin system. (a,b) Space-filling representations showing a top and side view, respectively, of the crystal structure of a double mutant form of GroEL (26). Two adjacent subunits are colored with the apical domains in red and purple, the intermediate domains in orange and yellow, and the equatorial domains in blue and green, respectively. Free passage between the GroEL rings is obstructed by N- and C-terminal residues not resolved in the crystal structure. (b) side view of GroES (28) at the top; two adjacent domains and a single mobile loop that is structured in the GroES crystal due to crystal packing are colored. The loop regions normally protrude from the base of GroES downward toward the GroEL. (c) Asymmetrical GroEL/GroES complexes as revealed by cryoelectron microscopy (45). Note the upward and outward movement of the apical GroEL domains interacting with GroES. (d) Model of the GroEL/GroES reaction cycle in assisting protein folding (30); see text for details. The term unfolded protein refers to a partially folded intermediate that is represented by the light pink spheres; folded protein is represented by the dark pink sphere, while the hatched spheres represent a mixture of folded and partially- folded proteins expected in a population of GroEL molecules. At step 4, GroES may associate with either the protein-containing ring of GroEL or with the opposite empty ring; the latter possibility is not shown. Reprinted from Nature 381, 571–580 (1996), with permission; copyright (1996) Macmillan Magazines Ltd.

Figure 1. (continued) Structure and function of the chaperonin system. (a,b) Space-filling representations showing a top and side view, respectively, of the crystal structure of a double mutant form of GroEL (26). Two adjacent subunits are colored with the apical domains in red and purple, the intermediate domains in orange and yellow, and the equatorial domains in blue and green, respectively. Free passage between the GroEL rings is obstructed by N- and C-terminal residues not resolved in the crystal structure. (b) side view of GroES (28) at the top; two adjacent domains and a single mobile loop that is structured in the GroES crystal due to crystal packing are colored. The loop regions normally protrude from the base of GroES downward toward the GroEL. (c) Asymmetrical GroEL/GroES complexes as revealed by cryoelectron microscopy (45). Note the upward and outward movement of the apical GroEL domains interacting with GroES. (d) Model of the GroEL/GroES reaction cycle in assisting protein folding (30); see text for details. The term unfolded protein refers to a partially folded intermediate that is represented by the light pink spheres; folded protein is represented by the dark pink sphere, while the hatched spheres represent a mixture of folded and partially- folded proteins expected in a population of GroEL molecules. At step 4, GroES may associate with either the protein-containing ring of GroEL or with the opposite empty ring; the latter possibility is not shown. Reprinted from Nature 381, 571–580 (1996), with permission; copyright (1996) Macmillan Magazines Ltd.
No crystal structure for any TCP-1 chaperonin is yet available, but electron-microscopic studies suggest that the general architecture and overall domain structure of the TCP-1 cylinder resembles that of GroEL (33, 34). The archaebacterial chaperonin was originally termed thermophilic factor 55, because it is virtually the only protein made by thermophilic archaebacteria under heat-shock conditions (35), but it is now termed the thermosome (36). Thermosomes generally contain eight subunits per ring, but there are several reports that a significant subset of particles contain nine subunits per ring, while the chaperonins of some Sulfolobus species appear all to contain nine subunits per ring (37). The thermosomes of Pyrodictium, Thermoplasma, and Sulfolobus consist of two different but related subunits of almost identical molecular mass (33). Electron-microscopic image analysis suggests that the two types of subunit in the thermosome of Thermoplasma acidophilum alternate within each ring, generating a four-fold symmetry (38).
Averaged electron-microscopic images of the mammalian TCP-1 particle reveal a ringlike structure with eightfold quasi-rotational symmetry with a central channel (33). Primary structures of eight distinct types of related subunit have been determined by cloning and sequencing of mouse cDNA; these types share about 30% amino acid residue identity. A striking observation is that the sequence of each type is more highly conserved among different eukaryotic species than are the sequences between types in any one species; in mammals the sequence identity is over 96% for each type of subunit, and around 60% between yeast and mouse. These observations have prompted the conclusion that each type of subunit diverged early in evolution, has changed only slowly during the evolution of eukaryotes, and thus may have a specific function in binding subsets of sequences in substrate proteins and/or other molecular chaperones (6, 7). Consistent with this suggestion is the observation that the different types of subunit in each TCP-1 chaperonin ring deviate most from one another in the sequences of the apical domains, where the protein substrate binds.
4. Mechanism of Action
The molecular details of how the chaperonins function have, and continue to be, studied by means of in vitro protein refolding experiments using defined components, but such experiments suffer from the disadvantage that the conditions under which they are performed are different in important respects from those operating during protein folding in the intact cell (9, 18). Thus it is not surprising that this field is subject to intense debate between people supporting different views (39). Since the chaperonins evolved to fold proteins inside cells,and not inside test tubes, it is essential when evaluating the validity of these views to define those aspects of chaperonin action observed in vitro that are mechanistically essential to improve the yield of correctly folded protein, when extrapolated as far as possible to in vivo conditions. In our opinion, the model outlined in Figure 1d best meets this criterion for the GroE chaperonins (30, 40).
In this model, newly synthesized polypeptide chains are released from ribosomes in the form of partially folded intermediates that may contain bound molecular chaperones of the hsp70/DnaJ families. The latter chaperones prevent aggregation and premature folding before each chain is complete. Such intermediates are commonly referred to as unfolded proteins, but in fact they often are molten globules. These partially folded, compact intermediates lose the hsp70/DnaJ chaperones by an uncharacterized mechanism that results in each intermediate molecule binding by hydrophobic interactions to the apical domains at one end of a GroEL cylinder; the other end of the cylinder is occupied by GroES as a result of ADP binding to the GroEL ring proximal to the GroES. The binding of polypeptide and the hydrolysis of ATP by the ring not occupied by GroES causes the GroES and the ADP to dissociate (step 2). GroES and ATP then rebind with equal probability to either ring (not both rings), resulting in 50% of the bound polypeptide being encapsulated inside the cavity capped by GroES (step 4). This rebinding of GroES results in the displacement of the bound polypeptide into the cavity, where it can continue to fold; it is for this reason that this model has been dubbed the ”Anfinsen cage” model to stress the idea that the protein in the cavity folds in a similar, but not necessarily identical, manner to that by which it folds in a protein renaturation experiment of the type pioneered by Anfinsen (41, 42). Thus one essential difference between chaperonin-assisted protein folding in vivo and spontaneous protein refolding in vitro is that the former process segregates each chain into the protected compartment provided by the GroEL/GroES cylinder to avoid the problem of aggregation.
The time available for this protected folding is set by the time it takes each ring of GroEL to hydrolyze seven ATP molecules. This hydrolysis is positively cooperative in each ring, and two rounds take about 10 s at 37°C (steps 4 and 5). The GroES then dissociates, and the protein is free either to diffuse into the cytosol or to rebind to the apical domains of the same GroEL cylinder (step 6). Rebinding occurs only if sufficient hydrophobic residues are still exposed on the surface of the compact intermediate. The cycle of release into the cage then repeats until the protein is folded sufficiently to the point where aggregation with other partially folded proteins is no longer a problem for the cell. The number of reaction cycles will vary, depending on the rate at which a given protein can fold when released into the cavity. Even with a single type of protein substrate, the population of folding chains will not be in synchrony or occupy the same number of cycles, since some chains fold more rapidly than others of the same sequence. Usually only a fraction of the polypeptide chains fold to completion in a single cycle of GroES binding and unbinding, depending on the intrinsic rate of folding of a particular polypeptide and its tendency to become trapped in misfolded states. Rebinding of these intermediates to the apical domains of GoEL results in partial unfolding in preparation for a subsequent folding trial (30). This unfolding function distinguishes the GroEL/ES machinery from a simple folding cage,which acts solely to prevent intermolecular aggregation.
Uncertainties about this model include the fate of the 50% of polypeptide bound to the GroEL ring not capped by GroES; such polypeptides would be prone to aggregation and degradation if released into the cytosol. Release of such polypeptides in vitro is reduced in the presence of high concentrations of synthetic polymers that mimic the macromolecular crowding, excluded volume effect present in the cytosol (40). However, some release of partially folded chains may be essential in vivo because of the danger that proteins that are unable to fold correctly as a result of mutation will clog up the GroEL apical domains; an escape route for such molecules may be provided by release from the ring not capped by GroES.
Another problem concerns the significance of GroEL molecules with GroES bound to each end of the cylinder that have been observed in vitro (43). The ratio of total GroES subunits to total GroEL subunits in extracts of E. coli cells is equimolar, consistent with such GroES/GroEL/GroES complexes occurring in vivo, where their formation may be favored by the effect of macromolecular crowding on protein association. Nevertheless, such complexes are not mechanistically essential, at least as deduced from in vitro refolding experiments, nor can they be permanent if the compact intermediates are to enter the GroEL cavities.
Another limitation imposed by this model is the size range that can be accommodated inside the cage, which is likely to have an upper limit around 60 kDa. Larger proteins than this, up to about 150 kDa, exist inside bacterial cells and even larger ones in the cytosol of eukaryotic cells. A GroES equivalent to cap the TCP-1 chaperonin oligomer has not been identified, so it is possible that the latter assists protein folding in a manner somewhat different from that for the GroE chaperonins. There is evidence that, unlike GroEL,the mammalian TCP-1 chaperonin binds to chains of actin being synthesized by extracts of reticulocytes before the chains have been released from the ribosomes (44).
There are four known intracellular compartments in which proteins fold that do not appear to contain any type of chaperonin: the endoplasmic reticulum lumen, the intermembrane mitochondrial space, the intrathylakoidal lumen, and the periplasmic space of Gram-negative bacteria. Additionally, the eukaryotic cytosol seems to lack a general chaperonin, since the TCP-1 chaperonin appears to be restricted to a small subset of protein substrates, including actin and tubulin. Thus it is certainly not the case that the folding of all proteins in all cells involves the chaperonins. Given that the intracellular environment strongly favors aggregation, it is likely that the chaperonin-independent proteins utilize the assistance of other types of molecular chaperone for their folding. This assistance may also apply to proteins larger than 60 kDa that cannot fit into the chaperonin cage. For these proteins, the cotranslational and sequential folding of protein domains may circumvent the requirement for a sequestered folding compartment.
It is also possible that some chaperonins have roles additional to those discussed above with respect to protein folding, since there are sporadic reports of chaperonin-like molecules occurring on the cell surface in both prokaryotic and eukaryotic cells and in the blood serum, as well as stimulating cytokine production by animal cell cultures. The bacterial chaperonins are also the major immunogens in all human bacterial infections, and their possible roles in protective immunity and autoimmune disease is an active area of research (45).
5. Note Added in Proof
The cage mechanism for chaperonin-assisted protein folding is strongly supported by recent structural and functional data. The crystal structure of the asymmetrical GroEL:GroES complex with bound ADP shows that the cage is sufficient to accept proteins up to at least 70 kDa and that the wall of the cage provides a hydrophilic environment permissive for folding. Crystal structures of the archaean thermosome suggest that in this chaperonin the folding cage is closed by loop sequences that emanate from the apical domains of the chaperonin subunits, explaining the lack of a separate GroES-like factor (M. Klumpff et al. Cell 91, 263–270, 1997; L. Ditzel et al. Cell 93, 125–138, 1998). A single ring mitochondrial chaperonin is fully functional in protein folding in a GroES-dependent manner in vivo (K. L. Nielsen et al. Mol. Cell 2, 93–99, 1998). The size range of GroEL substrate proteins and their kinetics of interaction in vivo are consistent with the cage mechanism (K. L. Ewalt et al. Cell 90, 491–500, 1997). Oligomeric GroEL with an intact central cavity is essential for the maintenence of growth of E. coli (F. Weber et al. Nature Struct. Biol. 11, 977–985, 1998).
References
1. S. M. Hemmingsen, C. Woolford, S. M. van der Vies, K. Tilly, D. T. Dennis, G. C. Georgopoulos, R. W. Hendrix, and R. J. Ellis (1988) Nature 333, 330–334.
2. R. S. Gupta (1990) Biochem. Int. 20, 833–841.
3. R. J. Ellis (1990) Science 250, 954–959.
4. C. Georgopoulos, D. Ang, K. Liberek, and M. Zylicz (1990) in R. I. Morimoto, A. Tissieres, and C. Georgopoulos, eds., Stress Proteins in Biology and Medicine, Cold Spring Harbor Laboratory Press, New York, pp. 191–221.
5. L. Silver, K. Artzt, and D. Bennett (1979) Cell 17, 275–284.
6. H. Kubota, G. Hynes, and K. Willison (1995) Eur. J. Biochem. 230, 3–16.
7. K. R. Willison and A. L. Horwich (1996) in R. J. Ellis, ed., The Chaperonins, Academic Press, San Diego, pp. 108–136.
8. F. U. Hartl (1996) Nature 381, 571–580.
9. R. J. Ellis and F. U. Hartl (1996) FASEB J. 10, 20–26.
10. E. M. Hallberg, Y. Shu, and R. L. Hallberg (1993) Mol. Cell Biol. 13, 3050–3057.
11. A. L. Horwich, K. B. Low, F. A. Fenton, I. N. Hirshfield, and K. Furtak (1993) Cell 74, 909–917 .
12. P. Goloubinoff, J. P. Christeller, A. A. Gatenby, and G. H. Lorimer (1989) Nature 342, 884–889.
13. J. Buchner, M. Schmidt, M. Fuchs, R. Jaenicke, R. Rudolph, F. X. Schmid, and T. Kiefhaber
(1991) Biochemistry 30, 1586–1591.
14. J. Martin, T. Langer, R. Boteva, A. Schramel, A. L. Horwich, and F.-U. Hartl (1991) Nature 352, 36-42.
15. G. S. Jackson, R. A. Staniforth, D. J. Halsall, T. Atkinson, J. J. Holbrook, A. R. Clarke, and S. G. Burston (1993) Biochemistry 32, 2554–2563.
16. J. Martin, A. L. Horwich, and F. U. Hartl (1992) Science 258, 995–998.
17. A. Guagliardi, L. Cerchia, S. Bartolucci, and M. Rossi (1994) Protein Sci. 3, 1436–1443.
18. R. J. Ellis (1996) in R. J. Ellis, ed., The Chaperonins, Academic Press, San Diego, pp. 1–25.
19. D. Ursic, J. C. Sedbrook, K. L. Himmel, and M. R. Culbertson (1994) Mol. Biol. Cell 5, 1065–1080.
20. H. Sternlicht, G. W. Farr, M. L. Sternlicht, J. K. Driscoll, K. R. Willison, and M. B. Yaffe (1993) Proc. Natl. Acad. Sci USA 90, 9422–9426.
21. R. S. Gupta (1996) in R. J. Ellis, ed., The Chaperonins, Academic Press, San Diego, pp. 27–64.
22. U. Bertsch, J. Soll, R. Seetharam, and P. V. Viitanen (1992) Proc. Natl. Acad. Sci. USA 89, 8696-8700.
23. K. F. Liou and K. R. Willison (1997) EMBO J. 16, 101–106.
24. A. M. Roseman, S. Chen, H. White, K. Braig, and H. R. Saibil (1996) Cell 87, 241–251.
25. O. Llorca, S. Marco, J. L. Carrascosa, and J. M. Valpuesta (1997) J. Struct. Biol. 118, 31–42.
26. K. Braig, Z. Otwinowski, R. Hegde, D. C. Boisvert, A. Joachimiak, A. L. Horwich, and P. B. Sigler (1994) Nature 371, 578–586.
27. S. C. Mande, V. Mehra, B. Bloom, and W. G. J. Hol (1996) Science 271, 203–207.
28. J. F. Hunt, A. J. Weaver, S. J. Landry, L. Gierasch, and J. Diesenhofer (1996) Nature 379, 37–42.
29. J. Martin, M. Mayhew, T. Langer, and F. U. Hartl (1993) Nature 366, 228–233.
30. M. Mayhew, A. C. R. da Silva, J. Martin, H. Erdjument-Bromage, P. Tempst, and F. U. Hartl (1996) Nature 379, 420–426.
31. J. S. Weissman, H. S. Rye, W. A. Fenton, J. M. Beecham, and A. L. Horwich (1996) Cell 84, 481-490.
32. S. Chen, A. M. Roseman, A. S. Hunter, S. P. Wood, S. G. Burston, N. A. Ranson, A. R. Clarke, and H. R. Saibil (1994) Nature 371, 261–264.
33. T. Waldmann, E. Nimmesgern, M. Nitsch, J. Peters, G. Pfeifer, S. Muller, J. Kellermann, A. Engel, F. U. Hartl, and W. Baumeister (1995) Eur. J. Biochem. 227, 848–856.
34. V. A. Lewis, G. M. Hynes, D. Zheng, H. Saibil, and K. Willison (1992) Nature 358, 249–252.
35. B. M. Phipps, A. Hoffmann, K. O. Stetter, and W. Baumeister (1991) EMBO J. 10, 1711–1722.
36. B. M. Phipps, D. Typke, R. Hegerl, S. Volker, A. Hoffmann, K. O. Stetter, and W. Baumeister (1993) Nature 361, 475–477.
37. S. Marco, D. Urena, J. L. Carrascosa, T. Waldmann, J. Peters, R. Hegerl, G. Pfeifer, H. Sack-Kongehl, and W. Baumeister (1994) FEBS Lett. 341, 152–155.
38. M. Nitsch, M. Klumpp, A. Lupas, and W. Baumeister (1997) J. Mol. Biol. 267, 142–149.
39. A. C. Clarke and P. A. Lund (1996) R. J. Ellis, ed., in The Chaperonins Academic Press, San Diego, pp. 168–212.
40. J. Martin and F. U. Hartl (1997) Proc. Natl. Acad. Sci. USA 94, 1107–1112.
41. R. J. Ellis (1994) Curr. Biol. 4, 633–635.
42. R. J. Ellis (1996) Fold. Des. 1, R9–R15.
43. Z. Torok, L. Vigh, and P. Goloubinoff (1996) J. Biol. Chem. 271, 16180–16186.
44. J. Frydman and F. U. Hartl (1996) Science 272, 1497–1502.
45.A. R. M. Coates (1996) in R. J. Ellis, ed., The Chaperonins Academic Press, San Diego, pp. 268-296.

|
|
|
|
لخفض ضغط الدم.. دراسة تحدد "تمارين مهمة"
|
|
|
|
|
|
|
طال انتظارها.. ميزة جديدة من "واتساب" تعزز الخصوصية
|
|
|
|
|
|
|
مشاتل الكفيل تزيّن مجمّع أبي الفضل العبّاس (عليه السلام) بالورد استعدادًا لحفل التخرج المركزي
|
|
|