آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Eukaryotic Protein-Coding Genes Can Be Identified by the Conservation of Exons and of Genome Organization |


|
|
Read More
Date: 15-11-2020
Date: 5-3-2021
Date: 6-11-2020
|
Eukaryotic Protein-Coding Genes Can Be Identified by the Conservation of Exons and of Genome Organization
KEY CONCEPTS
- Researchers can use the conservation of exons as the basis for identifying coding regions as sequences that are present in multiple organisms.
- Methods for identifying functional genes are not perfect and many corrections must be made to preliminary estimates.
- Pseudogenes must be distinguished from functional genes.
- There are extensive syntenic relationships between the mouse and human genomes, and most functional genes are in a syntenic region.
Some major approaches to identifying eukaryotic protein-coding genes are based on the contrast between the conservation of exons and the variation of introns. In a region containing a gene whose function has been conserved among a range of species, the sequence representing the polypeptide should have two distinctive properties:
1. It must have an open reading frame.
2. It is likely to have a related (orthologous) sequence in other species.
Researchers can use these features to identify functional genes. After we have determined the sequence of a genome, we still need to identify the genes within it. Coding sequences represent a very small fraction of the total genome. Potential exons can be identified as uninterrupted ORFs flanked by appropriate sequences. What criteria need to be satisfied to identify a functional (intact) gene from a series of exons?
FIGURE 1. shows that a functional gene should consist of a series of exons in which the first exon (containing an initiation codon) immediately follows a promoter, the internal exons are flanked by appropriate splicing junctions, and the last exon has the termination codon and is followed by 3′ processing signals; therefore, a single ORF starting with an initiation codon and ending with a termination codon can be deduced by joining the exons together. Internal exons can be identified as ORFs flanked by splicing junctions. In the simplest cases, the first and last exons contain the beginning and end of the coding region, respectively (as well as the 5′ and 3′ untranslated regions). In more complex cases, the first or last exons might have only untranslated regions and can therefore be more difficult to identify.
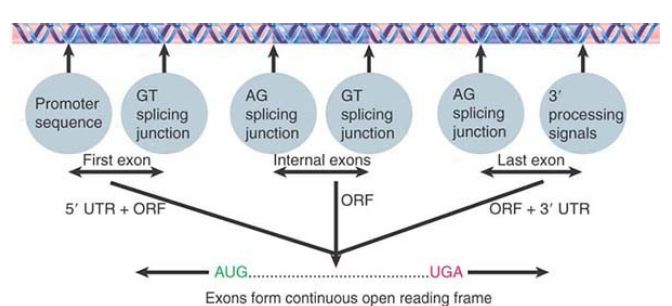
FIGURE 1. Exons of protein-coding genes are identified as coding sequences flanked by appropriate signals (with untranslated regions at both ends). The series of exons must generate an ORF with appropriate initiation and termination codons.
The algorithms that are used to connect exons are not completely effective when the gene is very large and the exons might be separated by very large distances. For example, the initial analysisof the human genome mapped 170,000 exons into 32,000 genes. This is incorrect because it gives an average of 5.3 exons per gene, whereas the average of individual genes that have been fully characterized is 10.2. Either we have missed many exons, or they should be connected differently into a smaller number of genes in the entire genome sequence.
Even when the organization of a gene is correctly identified, there is the problem of distinguishing functional genes from pseudogenes. Many pseudogenes can be recognized by obvious defects in the form of multiple mutations that result in nonfunctional coding sequences. Pseudogenes that have originated more recently have not accumulated so many mutations and thus may be more difficult to identify. In an extreme example, the mouse has only one functional encoding glyceraldehyde phosphate dehydrogenase gene (GAPDH), but has about 400 homologous pseudogenes.
Approximately 100 of these pseudogenes initially appeared to be functional in the mouse genome sequence, and individual examination was necessary to exclude them from the list of
functional genes. Pseudogenes with relatively intact coding sequences but mutated transcription signals are more difficult to identify.
How can suspected protein-coding genes be verified? If it can be shown that a DNA sequence is transcribed and processed into a translatable mRNA, it is assumed that it is functional. One technique for doing this is reverse transcription polymerasechain reaction (RT-PCR) , in which RNA isolated from cells is reverse transcribed to DNA and subsequently amplified to many copies using the polymerase chain reaction. The amplified DNA products can then be sequenced or otherwise analyzed to see if they have the appropriate structural features of a mature transcript.
RT-PCR can also be used for quantitative assessment of gene expression, although there are now better techniques for this purpose. High throughput sequencing of reverse-transcribed RNAs from a cell sample (known as deep RNA sequencing or RNA-seq) allows rapid analysis and quantitation of the sample’s transcriptome. The application of this technique to the genetic
model organisms Drosophila and C. elegans has revealed details about gene expression across the genome and the characterization of regulatory networks during development. Confidence that a gene is functional can be increased by comparing regions of the genomes of different species. There has been extensive overall reorganization of sequences between the mouse and human genomes, as seen in the simple fact that there are 23 chromosomes in the human haploid genome and 20 chromosomes in the mouse haploid genome. However, at the level of individual chromosomal regions, the order of genes is generally the same: When pairs of human and mouse homologs are compared, the genes located on either side also tend to be
homologs. This relationship is called synteny.
FIGURE 2. shows the relationship between mouse chromosome 1 and the human chromosomal set. Twenty-one segments in this mouse chromosome that have syntenic counterparts in human chromosomes have been identified. The extent of reshuffling that has occurred between the genomes is shown by the fact that the segments are spread among six different human chromosomes.
The same types of relationships are found in all mouse chromosomes except for the X chromosome, which is syntenic only with the human X chromosome. This is explained by the fact that the X is a special case, subject to dosage compensation to adjust for the difference between the one copy of males and the two copies of females (see the chapter titled Epigenetics II). This restriction can apply selective pressure against the translocation of genes to and from the X chromosome.
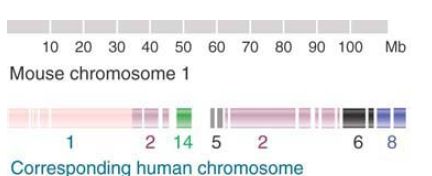
FIGURE 2. Mouse chromosome 1 has 21 segments between 1 and 25 Mb in length that are syntenic with regions corresponding to parts of six human chromosomes.
Comparison of the mouse and human genome sequences shows that more than 90% of each genome lies in syntenic blocks that range widely in size from 300 kb to 65 megabases (Mb). There is a total of 342 syntenic segments, with an average length of 7 Mb (0.3% of the genome). Ninety-nine percent of mouse genes have a homolog in the human genome; for 96% that homolog is in a syntenic region.
Comparison of genomes provides interesting information about the evolution of species. The number of gene families in the mouse and human genomes is the same, and a major difference between the species is the differential expansion of particular families in the mouse genome. This is especially noticeable in genes that affect phenotypic features that are unique to the species. Of 25 familiesfor which th e size has been expanded in the mouse genome, 14 contain genes specifically involved in rodent reproduction, and 5contain genes specific to the immune system.
A validation of the importance of the identification of syntenic blocks comes from pairwise comparisons of the genes within them. For example, a gene that is not in a syntenic location (i.e., its context is different in the two species being compared) is twice as likely to be a pseudogene. Put another way, gene translocation away from the original locus tends to be associated with the formation of pseudogenes. Therefore, the lack of a related gene in a syntenic position is grounds for suspecting that an apparent gene might really be a pseudogene. Overall, more than 10% of the genes that are initially identified by analysis of the genome are likely to turn out to be pseudogenes.
As a general rule, comparisons between genomes add significantly to the effectiveness of gene prediction. When sequence features indicating functional genes are conserved—for example, between human and mouse genomes—there is an increased probability that they identify functional orthologs.
Identifying genes encoding RNAs other than mRNA is more difficult because researchers cannot use the criterion of the ORF. It is certainly true that the comparative genome analysis described earlier has increased the rigor of the analysis. For example, analysis of either the human or the mouse genome alone identifies about 500 genes encoding tRNAs, but comparison of their features suggests that fewer than 350 of these genes are in fact functional in each genome.
Researchers can locate a functional gene through the use of an expressed sequence tag (EST), a short portion of a transcribed sequence usually obtained from sequencing one or both ends of a
cloned fragment from a cDNA library. An EST can confirm that a suspected gene is actually transcribed or help identify genes that influence particular disorders. Through the use of a physical mapping technique such as in situ hybridization (see the Clusters and Repeats chapter), researchers can determine the chromosomal location of an EST. (In situ hybridization is a technique that identifies the chromosomal location of a specific DNA sequence. We also can use it to determine the number of copies of a sequence in a cell, so it can detect whether there is an abnormal number of a specific chromosome. In this way, it is helpful in identifying cancerous cells, which often have extra copies of some chromosomes. It is also commonly used to diagnose suspected genetic disorders.)

|
|
|
|
دور في الحماية من السرطان.. يجب تناول لبن الزبادي يوميا
|
|
|
|
|
|
|
العلماء الروس يطورون مسيرة لمراقبة حرائق الغابات
|
|
|
|
|
|
|
ضمن أسبوع الإرشاد النفسي.. جامعة العميد تُقيم أنشطةً ثقافية وتطويرية لطلبتها
|
|
|