
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| F Plasmid |


|
|
Read More
Date: 28-10-2020
Date: 16-12-2015
Date: 10-12-2015
|
F Plasmid
In 1953 a new type of genetic element was postulated in order to account for the sexual behavior of Escherichia coli K-12 (1, 2). This determinant spread rapidly and independently through recipient cell populations, indicating considerable autonomy, but it was completely dispensable and conferred no phenotype on its host other than maleness. Nevertheless, it was stably inherited by the cells it invaded. It behaved, in essence, like a small, infectious chromosome, and it was baptized the fertility ) F) or sex factor. The nature of the proposed new element was amply confirmed: F turned out to be the prototype of the circular, double-stranded DNA plasmid.
F was immensely important to the development of molecular genetics. By promoting transfer of chromosomal genes, it opened the way to large-scale mapping of the bacterial genome; and by acquiring and transferring small portions of the host chromosome, it could create partial diploid conditions that enabled analyses of bacterial gene function and control of gene expression. The nature and consequences of F-specified maleness are generally understood and are described in the entry Hfr's and F-primes. F has also been at the forefront of investigations into how low-copy number replicons are maintained stably; the mechanisms that regulate F's maintenance will be the major focus here.
Early studies of F were greatly aided by the discovery of derivatives that carry chromosomal genes ( F-primes). For example, isolation of F′ lac+ mutants (in a lac mutant strain) that remained Lac+ at° 30C but segregated Lac– at 42°C demonstrated that F carries its own replication determinants (3). The selective inhibition of F maintenance by acridine dyes (4) also pointed to the autonomy of F replication. Measurements of b-galactosidase in F–/lac+ and F′lac+/lac+ strains gave the first indication that the copy number of F is about the same as that of the chromosome. The strict regulation of copy number was reflected in the phenomenon of incompatibility, the inability of two F plasmids (eg F′lac and F′gal) to be coinherited. The F replication system enables integrated F to assume control of chromosome replication in strains unable to initiate it themselves, a phenomenon known as “integrative suppression” (5, 6). Beyond the initiation of replication at the origin, however, F replication becomes strictly dependent on host functions. The term “episome” was coined to denote plasmids like F with both autonomous and passive (integrated) modes of propagation. This property turned out not to reflect a fundamental distinction from other plasmids, and the term is no longer considered useful.
1. Anatomy of F, A Mosaic
F consists of four functional blocks and a region of unknown composition (Fig. 1). The 33-kbp tra operon encodes the essential components of donor ability, the mating apparatus and the DNA manipulation enzymes responsible for transmitting F into recipient cells, as shown by the self-transmissibility acquired by plasmid vectors into which it, along with the origin of transfer (oriT), has been inserted. The ~ 13-kbp leading region that follows oriT is the first to enter recipient cells during transfer; only a few genes within it are known, and their involvement in transfer has not been demonstrated. The region at the other end of the tra operon contains a defunct replicon and appears to have acted as a trap for transposable elements; the latter contribute indirectly to mating activity, because one of them is inserted in the finO regulator gene, causing constitutive expression of the tra operon and high transfer rates, and (ii) also because homologous recombination with similar elements in the chromosome is the major route of F integration to form Hfr donors. The fourth region carries all the significant determinants of F maintenance. When isolated and joined to an antibiotic resistance gene to permit selection, it behaves as a normal plasmid (mini-F) with the same replication, partition, and stability properties as its parent F (7, 8). It is, therefore, the form most often used for studies of F maintenance.
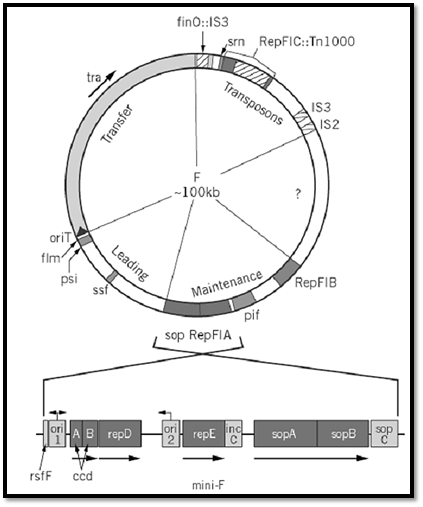
Figure 1. Genetic map of the F plasmid. The circular F plasmid has been divided into five sectors, roughly according to function. Gene symbols are explained in the text. The tra arrow indicates the direction of transcription of the tra operon. The oriT arrow indicates the direction of conjugal transfer. Unshaded blocks of the circle correspond approximately to unsequenced regions. Below is the repFIA mini-F; note that it is shown inverted relative to F, by convention. Arrows above ori1 and ori2 show bi- and unidirectional replication, respectively.
A closer view reveals F to be a mosaic of replicative determinants. Three have been identified: repFIA, repFIB, and repFIC. The first two can be isolated as miniplasmids, although replication of repFIB is partly defective, so that replication of F itself is predominantly governed by repFIA. The repFIC locus, identified as such by comparison with the rep sequences of other plasmids, is inactive due to insertion of Tn1000 in a progenitor of F. Portions of F as yet unsequenced may prove to contain other replicons. The presence of multiple replicons has proved to be a common feature of large plasmids, and it suggests that many such plasmids may have arisen by the fusion and rearrangement of a limited number of ancestral plasmids (9, 10). The fact that only repFIA is fully functional may account for the fact that F is stably maintained in only a narrow range of hosts: E. coli and similar Enterobacteria.
1.1. RepFIA
The repFIA locus is itself an ensemble of essential and accessory maintenance functions (Fig. 1.( Electron microscopic examination of replication intermediates showed that replication forks emanated bidirectionally from a site termed ori1 [also called oriV (11)]. However, miniplasmid deletion derivatives that lack this site, but show similar replication behavior, were obtained readily (12) . These replicate unidirectionally from ori2 [oriS (13)], both in vivo and in vitro (14, 15). It is not known why the ori1 site is preferred in the original mini-F. In any event, the smaller mini-F, replicating from ori2, has been used in most investigations.
The basic repFIA replicon is made up of three components. One is ori2. The others are the single essential replication gene, repE, whose product regulates all aspects of initiation, and the copy control region, incC (13, 16, 17). A plasmid comprising only these elements replicates normally, but is nevertheless unstable. Stable inheritance requires a mechanism to ensure that plasmid copies are directed into each of the daughter cells, and this function is provided by the adjacent sop locus (18). Sop is essentially a partition module that can act independently of the rep locus to which it is normally linked, to stabilize plasmid vectors in which it is inserted. Together, the rep and sop loci constitute the basic F maintenance system. This arrangement is remarkably similar to that found in the P1 prophage plasmid [a detailed level, RepFIB is probably a more exact analogue of the mini-P1 replicon]. Studies of mini-F and mini-P1 have been reciprocally informative.
2. F in the Cell Cycle
To ensure that F makes replicas available for both daughter cells at division, in spite of its low copy number, it would appear necessary for replication to be coordinated with the cell cycle. Viewed simply, this would imply that F replication take place at a specific time in the cycle for given growth conditions, as does the initiation of chromosome replication. However, a number of careful studies have indicated that both F and mini-F replicate in cells of all ages, without delaying cell division. Despite some dissenting opinion (21), it appears reasonable at present to view replication of F as more loosely coordinated with the cell cycle than that of the chromosome. How F could achieve high hereditary stability in the absence of cell cycle coordination remains unclear.
3. Replication Control
F provides the regulator of its replication, while the chromosome contributes essential components to the mechanism of initiation at ori2. Thus, the host DnaA initiator protein binds on one side of the origin to enhance opening of an AT-rich region (Fig. 2a). Initiation itself, however, requires binding of the F RepE protein to specific sites (iterons) on the other side (22), so that the duplex becomes sufficiently deformed to allow entry of DnaB helicase and assembly of a replication complex (23, 24).
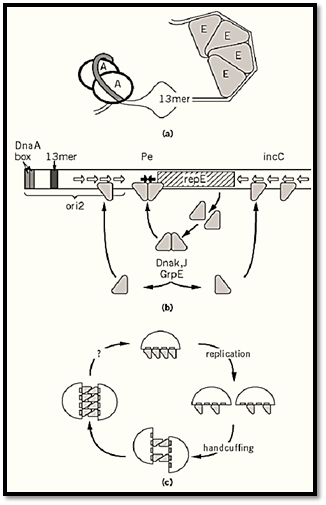
Figure 2. Aspects of mini-F replication. (a) Opening of ori2 at initiation; A is DnaA protein, bound at tandem DnaA boxes (shaded DNA), E is RepE monomer bound to the iterons of ori2; together they cause melting of the AT-rich region at a 13-mer sequence similar to those that are opened at an early stage of initiation in oriC. (b) Regulation of RepE synthesis and activity. Each arm of the operator (filled arrows) contains an 8-bp subsequence of the 19-mer iteron binding sites (open arrows), suggesting that initiation, negative control, and autoregulation all depend on interaction of the same RepE binding domain with DNA. Limiting RepE synthesis by autoregulated transcription, along with limiting RepE availability through binding to incC, could constitute a copy-control mechanism. But how can competitive binding be effective if an autoregulatory mechanism exists to replenish the pool of free RepE? One solution is that RepE exists in two forms: It is first synthesized as a repressor, which binds to the operator but not to the origin, and part of it is then converted to an initiator form that binds to incC and ori2 . In this way, titration and autoregulation become to a large extent independent of each other (30). Indeed, the complex formed by RepE with the operator is structurally distinct from that formed with ori2 (31). The model is further supported by the finding that most RepE is present in a dimer form with operator-binding and repressor activity. The initiator is made when the dimer is converted by the DnaK–DnaJ–GrpE chaperone machine to a monomer, which binds to the ori2 and incC iterons (29). These chaperones are thus needed for normal levels of mini-F replication (32). Certain mini-F mutants that have escaped chaperone-dependence make RepE that has lost the ability to dimerize, resulting in high copy numbers (29, 33). (c) Principle of handcuffing and the cycle of initiation and inhibition; the interactions shown are intermolecular, but intramolecular coupling is also possible. After replication, the RepE remaining at ori2 pairs with RepE remaining either at incC or at ori2 on a second plasmid. As more RepE is made, it also pairs, inhibiting initiation by hindering access to replication proteins or impeding the structural transition needed for initiation. Failure to pair is a feature of certain copy-mutant initiator proteins of plasmids P1, RK2, and R6K (35-37). An unknown event (question mark) ruptures the coupling to allow initiation.
The incC control region acts as a brake on the formation of RepE-ori2 initiation complexes. Additional copies of incC in trans inhibit replication, while deletion of incC causes about a fivefold increase in copy number (16, 17, 25). Because incC binds RepE (Fig. 2b), it was easy to imagine that it competes with ori2 for RepE, controlling replication by titrating the initiator. An alternative copy-control mechanism sprang from studies of plasmids P1 and RK2 (26, 27), in which it was found that initiators bound to iteron sites can also bind to each other. The iteron regions ori2 and incC could thus pair, either intra- or intermolecularly, to block initiation. This model, termed “handcuffing,” would explain regulation of the frequency of initiation by a dual activity of RepE: initial reinforcement of the handcuff to prevent initiation, followed by formation of an initiation complex to
start replication (Fig. 2c). A subtle modulation of RepE synthesis would seem necessary to allow the transition.
In fact, production of RepE initiator is finely regulated. First, RepE represses transcription of its own gene, by binding to the repE promoter (28), accounting for the limited quantities of RepE initiator. Second, newly made RepE has only this activity and is unable to initiate replication. This is because newly synthesized RepE forms dimers, which bind to the inverted repeat operator sequence in the repE promoter region but not to the iterons in ori2. To become an initiator, RepE must be converted to a monomer form (29-31). This conversion is catalyzed by the DnaK-DnaJ -GrpE molecular chaperone machine, a host function that is needed for normal levels of mini-F replication (29, 32, 33).
Despite the identification of these elements of F replication control, a satisfactory explanation of this process is still beyond us. The discovery of the essential role of the chaperone machine has served only to push the question of the key regulatory process one step further back: What determines the rate at which RepE dimer is converted to active monomer? If the handcuffing model is correct, how is the pairing broken to allow replication? Motor forces of the partition system have been proposed as keys (or boltcutters) for removing the handcuffs (34). But if handcuffed plasmids are pulled apart by the partition apparatus, how does unpairing occur in the case of partition-defective mini-F mutants, which replicate at a normal rate? The true status of ori1 is unclear: If it is really the major origin in wild-type F, why is DnaA-assisted strand-opening over 2 kbp away essential for initiation?
4. Partition
Deletion of the sop region causes loss of mini-F at the rate expected for random distribution of a small number of plasmid copies to daughter cells prior to division, consistent with the disruption of an active partitioning mechanism. The sop region consists of three elements (Fig. 1), all essential for partition (18, 38). Two partition proteins, SopA and SopB, are encoded by the sop operon. SopA is the main regulator of the operon; it binds to a series of short repeated sequences in the promoter to repress transcription (39). SopB may also assist this repression, because in vitro it enhances the affinity of SopA for its promoter (39). The major role of SopB, however, is the formation of a partition complex with sopC, a series of 12 tandem repeats of 43 bp that act in cis to ensure partition, much like a eukaryotic centromere. SopB appears to bind as a dimer to an inverted repeat sequence in each 43-mer, and to wrap sopC DNA in positive supercoils to form a characteristic complex (40, 41).
How this complex acts in partition is still unknown. Current models suppose that plasmid replicas attach as paired molecules to a host structure, such as the membrane or the division septum, and that some force then splits the pairs, pulling one copy toward each pole. SopA has an ATPase activity and is presumed to have a function in partition beyond its regulatory activity. It may link the SopB-sopC complex to the host structure, where its ATPase activity could drive the separation of paired plasmid copies (42). Our ignorance of the host components with which the Sop complex interacts is presently the major barrier to understanding partition. Host mutations that influence mini-F stability have been located in the gyrA, topA, and ugpA genes (43-45). While the DNA topology function or membrane localization of these proteins is suggestive, there is as yet no good explanation for their roles in partition.
Recent refinements in cytochemical methods applied to bacterial cells have made possible the localization of plasmid molecules as fluorescent foci, allowing the movement of mini-F and other plasmids to be followed in individual cells and populations (20, 46). The data thus far published indicate a preferential location at mid-cell (the site of the next division septum) for mini-F (Fig. 3). After the focus doubles, presumably reflecting replication of the plasmid, the two new foci appear to move to the 1 / 4 and 3 / 4 positions, the midpoints of the emerging daughter cells, at a speed suggestive of an active, directed mechanism. Partition-defective mini-F shows a more random distribution and is often present close to the cell poles (20). These results are consistent with old observations of the paucity of sop+ F plasmids in anucleate cells (“ minicells”) created by division near the cell poles and with the contrasting abundance of sop mutant plasmids (47).
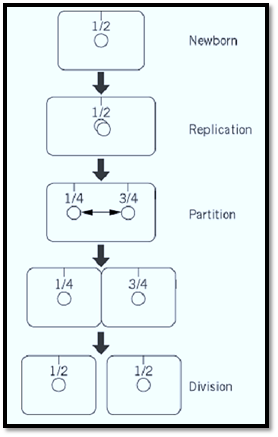
Figure 3. Positioning and displacement of mini-F during the cell cycle, based on microscopic examination of cells labeled with fluorescent probes (20, 46). The 1 / 4 and 3 / 4 positions in older cells become the 1 / 2 positions in new cells. Note that the smallness of the interval between the appearance of two mini-Fs and their appearance at the new cell mid-poles implies active displacement (double-headed arrow).
5. Resolution
The stability of DNA species that replicate as q-shaped molecules is under continual threat from the formation of dimers by homologous recombination, because this decreases the number of separate molecules available for partition and reduces initiation frequency in dimer-carrying progeny. Most such replicons have solved this problem by acquiring a site-specific recombination system that resolves dimers back to monomers. RepFIA is one of these. The product of the resD gene is necessary for the resolution, which takes place within a sequence containing an inverted repeat of 10 bp, the rfsF site (48, 49). ResD is presumed to be the resolvase, though in vitro evidence is lacking.
6. Addiction
F possesses another type of stability function that, instead of ensuring that each daughter cell inherits a plasmid copy, kills those that fail to. The host population is thus coerced into maintaining the plasmid, a situation likened to addiction (50). The ccd (controlled cell death) system turned out to be the first of many examples of a widespread “stable-toxin-unstable-antidote” strategy (51). The CcdB protein inhibits an essential cellular function, DNA gyrase, but in F+ cells it is kept in check by complexing with the relatively unstable antidote protein, CcdA (52, 53). In an F– segregant cell, the CcdA is lost by rapid proteolysis and cannot be replaced, and the CcdB liberated can attack gyrase and thereby kill the cell.
Two other toxin–antidote systems, srn and flm, are carried by F. SrnB promotes the degradation of ribosomal RNA and transfer RNA by damaging the inner membrane and allowing entry of ribonuclease I (54), but its synthesis is normally inhibited by an unstable anti-sense RNA, SrnC (55) . As with the Ccd system, loss of the plasmid causes a rapid decline in the antidote SrnC, and the liberated and stabilized srnB messenger RNA is then translated to produce the toxin. The locus is situated next to repFIC::Tn1000 and was undoubtedly once part of a viable replicon, similar to the R1 plasmid whose hok/ sok killer system it resembles. Flm is another hok/sok analogue. Its toxin ) flmA) and antidote RNA ( flmB) genes are located in the leading region (56). It is tempting to suggest that a burst of FlmA synthesis in the recipient modifies the membrane to facilitate conjugational transfer, but the actual relevance of flm is unknown.
Easy isolation of F– and mini-F– segregants suggests that these toxin–antidote systems are inefficient. This may, however, be the result of unintended laboratory selection for tolerant strains during decades of genetic experimentation. The significance of such addictive systems in the wild remains to be assessed.
7. Conjugal Aids
Two other loci in the leading region encode functions for which a role in conjugation can be envisaged. The psiB gene product is transiently expressed following transfer to a recipient cell, where it inhibits the SOS response, probably by preventing the activation of RecA protein (57). Its role might be to prevent induction of inappropriate SOS functions in the recipient. The ssf gene, an analogue of E. coli ssb, encodes a single-stranded DNA binding protein that might protect DNA entering the cell prior to its replication (58). The absence of leading region functions does not, however, impede DNA transfer or recombinant formation, at least in laboratory conditions, and their significance is thus unclear.
8. Phage Exclusion
Following infection of an F+ cell by certain phages dubbed “female-specific”, (eg, T7 and FII, (expression of phage genes begins and the cell dies, but phage development is arrested. The F locus responsible is pif (phage inhibition by F), an operon consisting of two genes, pifC and pifA, located between the repFIA and repFIB regions. The phenotype is not understood. It appears to be due largely to interactions between PifA and the products of phage genes 1.2 and 10, but it does not seem to be related to the normal activities of these genes. These interactions somehow interrupt entry of phage DNA and cut short expression of its late genes (59). PifC represses pif operon transcription by binding to a short inverted repeat sequence overlapping the pif promoter –10 region (60). The participation of PifC in mini-F replication initiated at ori1 has also been proposed, on the basis of the defective replication phenotypes of pifC amber mutants (61). It is also possible, however, that the phenotype is an indirect effect of PifC amber fragments binding to the pif promoter.
9. Perspectives
The F plasmid as a tool has largely retired from the front line of molecular genetics, to be replaced by sleeker, more powerful technologies. Nevertheless, its properties continue to be exploited, eg, its stability and low copy number make it suitable as a vector for cloning large fragments of eukaryotic DNA [the BAC vectors (62)]. As a biological entity, on the other hand, F has retained its mystery. We understand only dimly the control of its replication, its mechanism of partition, and the relationship of both processes to the host cell cycle, not only for F, but for all low-copy-number replicons. Finally, although whole-genome sequencing has not yet proved equal to the task of completing the sequence of F, this project is underway (R. Skurray and L. Frost, personal communications) and may well provide some surprises.
References
1. W. Hayes (1953) J. Gen. Microbiol. 8, 72–88.
2. L. L. Cavalli, J. Lederberg, and E. M. Lederberg (1953) J. Gen. Microbiol. 8, 89–103.
3. F. Cuzin and F. Jacob (1967) Ann. Inst. Pasteur 112, 397–418.
4.Y. Hirota (1960) Proc. Natl. Acad. Sci. USA 46, 57–64.
5. Y. Nishimura, L. Caro, C. M. Berg, and Y. Hirota (1971) J. Mol. Biol. 55, 441–456.
6. K. von Meyenberg and F. G. Hansen (1980) In Mechanistic Studies of DNA Replication and Genetic Recombination, ICN-UCLA Symposium on Molecular and Cellular Biology (B. Alberts and C. F. Fox, eds.), Academic Press, New York, pp. 137–159.
7. K. Timmis, F. Cabello, and S. N. Cohen (1975) Proc. Natl. Acad. Sci. USA 72, 2242–2246.
8. M. A. Lovett and D. R. Helinski (1976) J. Bacteriol. 127, 982–989.
9. D. Lane and R. C. Gardner (1979) J. Bacteriol. 139, 141–151.
10. P. L. Bergquist, H. E. D. Lane, L. Malcolm, and R. A. Doward (1982) J. Gen. Microbiol. 128, 223-238.
11. R. Eichenlaub, D. Figurski, and D. R. Helinski (1977) Proc. Natl. Acad. Sci. USA 74, 1138–1141.
12. J. J. Manis and B. C. Kline (1977) Mol. Gen. Genet. 152, 175–182.
13. D. Figurski, R. Kolter, R. Meyer, M. Kahn, R. Eichenlaub, and D. R. Helinski (1978) In Microbiology—1978 (D. Schlessinger, ed.), ASM, Washington, D.C., pp. 105–109.
14. T. Murotsu, H. Tsutsui, and K. Matsubara (1984) Mol. Gen. Genet. 196, 373–378.
15. K. Muraiso, T. Tokino, T. Murotsu, and K. Matsubara (1987) Mol. Gen. Genet. 206, 519–521.
16. P. L. Bergquist, R. A. Downard, P. A. Caughey, and H. E. D. Lane (1981) J. Bacteriol. 147, 888-899.
17. H. Tsutsui, A. Fujiyama, T. Murotsu, and K. Matsubara (1983) J. Bacteriol. 155, 337–344.
18. T. Ogura and S. Hiraga (1983) Cell 32, 351–360.
19. C. E. Helmstetter, M. Thornton, P. Zhou, J. A. Bogan, A. C. Leonard, and J. E. Grimwade (1997) J. Bacteriol. 179, 1393–1399.
20. H. Niki and S. Hiraga (1997) Cell 90, 951–957.
21. J. D. Keasling, B. O. Palsson, and S. Cooper (1992) Res. Microbiol. 143, 541–548.
22. T. Murotsu, K. Matsubara, H. Sugisaki, and M. Takanami (1981) Gene 15, 257–271.
23. D. Bramhill and A. Kornberg (1988) Cell 54, 915–918.
24. Y. Kawasaki, F. Matsunaga, Y. Kano, T. Yura, and C. Wada (1996) Mol. Gen. Genet. 253, 42–49.
25. H. Tsutsui and K. Matsubara (1981) J. Bacteriol. 147, 509–516.
26. S. K. Pal and D. K. Chattoraj (1988) J. Bacteriol. 170, 3554–3560.
27. M. J. McEachern, M. A. Bott, P. A. Tooker, and D. R. Helinski. (1989) Proc. Natl. Acad. Sci. USA 86, 7942–7946.
28. L. Sogaard-Andersen, L. A. Rokeach, and S. Molin (1984) EMBO J 3, 257–262.
29. M. Ishiai, C. Wada, Y. Kawasaki, and T. Yura (1994) Proc. Natl. Acad. Sci. USA 91, 3839–3843.
30. J. D. Trawick and B. C. Kline (1985) Plasmid 13, 59–69.
31. L. Masson and D. S. Ray (1986) Nucleic Acids Res. 14, 5693–5711.
32. Y. Kawasaki, C. Wada, and T. Yura (1990) Mol. Gen. Genet. 220, 277–282.
33. M. Ishiai, C. Wada, Y. Kawasaki, and T. Yura (1992) J. Bacteriol. 174, 5597–5603.
34. A. L. Abeles and S. J. Austin (1991) Proc. Natl. Acad. Sci. USA 88, 9011–9015.
35. G. Mukhopadhyay, S. Sozhamannan, and D. K. Chattoraj (1994) EMBO J 13, 2089–2096.
36. A. Blasina, B. L. Kittel, A. E. Toukdarian, and D. R. Helinski (1996) Proc. Natl. Acad. Sci. USA 93, 3559–3564.
37. A. Miron, I. Patel, and D. Bastia (1994) Proc. Natl. Acad. Sci. USA 91, 6438–6442.
38. H. Mori, A. Kondo, A. Ohshima, T. Ogura, and S. Hiraga (1986) J. Mol. Biol. 192, 1–15.
39. H. Mori, Y. Mori, C. Ichinose, H. Niki, T. Ogura, A. Kato, and S. Hiraga (1989) J. Biol. Chem. 264, 15535-15541.
40. D. P. Biek and J. Shi (1994) Proc. Natl. Acad. Sci. USA 91, 8027–8031.
41. A. S. Lynch and J. C. Wang (1994) J. Mol. Biol. 236, 679–684.
42. M. Motallebi-Veshareh, D. A. Roach, and C. M. Thomas (1990) Mol. Microbiol. 4, 1455–1463.
43. T. Ogura, H. Niki, H. Mori, M. Morita, M. Hasegawa, C. Ichinose, and S. Hiraga (1990) J. Bacteriol. 172, 1562–1568.
44. C. A. Miller, S. L. Beaucage, and S. N. Cohen (1990) Cell 62, 127–133.
45. B. Ezaki, H. Mori, T. Ogura, and S. Hiraga (1990) Mol. Gen. Genet. 223, 361–368.
46. G. S. Gordon, D. Sitnikov, C. D. Webb, A. Teleman, A. Straight, R. Losick, A. W. Murray, and A. Wright (1997) Cell 90, 1113–1121.
47. J. E. Hogan, B. C. Kline, and S. B. Levy (1982) Plasmid 8, 36–44.
48. D. Lane, R. de Feyter, M. Kennedy, S-H. Phua, and D. Semon (1986) Nucleic Acids Res. 14, 9713-9728.
49. M. B. O''Connor, J. J. Kilbane, and M. H. Malamy (1986) J. Mol. Biol. 189, 85–102.
50. H. Lehnherr, E. Maguin, S. Jafri, and M. B. Yarmolinsky (1993) J. Mol. Biol. 233, 414–428.
51. A. Jaffé, T. Ogura, and S. Hiraga (1985) J. Bacteriol. 163, 841–849.
52. P. Bernard and M. Couturier (1992) J. Mol. Biol. 226, 735–745.
53. S. Maki, S. Takiguchi, T. Miki, and T. Horiuchi (1992) J. Biol. Chem. 267, 12244–12251.
54. R. Ito and Y. Ohnishi (1983) Biochim. Biophys. Acta 20, 27–34.
55. A. K. Nielsen, P. Thorsted, T. Thisted, E. G. Wagner, and K. Gerdes (1991) Mol. Microbiol. 15, 1961-1973.
56. S. M. Loh, D. S. Cram, and R. A. Skurray (1988) Gene 66, 259–268.
57. M. Bagdasarian, A. Bailone, J. F. Angulo, P. Scholz, M. Bagdasarian, and R. Devoret (1992(Mol. Microbiol. 6, 885–893.
58. A. L. Kolodkin, M. A. Capage, E. I. Golub, and K. B. Low (1983) Proc. Natl. Acad. Sci. USA .80, 4422-4426.
59. C. K. Schmitt and I. J. Molineux (1991) J. Bacteriol. 173, 1536–1543.
60. J. F. Miller and M. H. Malamy (1986) Proc. Natl. Acad. Sci. USA 83, 1433–1437.

|
|
|
|
تفوقت في الاختبار على الجميع.. فاكهة "خارقة" في عالم التغذية
|
|
|
|
|
|
|
أمين عام أوبك: النفط الخام والغاز الطبيعي "هبة من الله"
|
|
|
|
|
|
|
خدمات متعددة يقدمها قسم الشؤون الخدمية للزائرين
|
|
|
















