
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Combinatorial Synthesis |


|
|
Read More
Date: 3-5-2021
Date: 8-12-2015
Date: 12-6-2021
|
Combinatorial Synthesis
The diversity of a molecular library may be derived from natural sources, or it may be created through specialized techniques in synthetic chemistry. Traditionally, synthetic chemists focus on preparing individual compounds, paying careful attention to stereochemical control and to achieving the greatest possible yield and purity. This often requires the use of highly specialized reactions and can be a painstakingly slow process. In contrast, chemists engaged in combinatorial organic synthesis employ transformations that are more general in scope and can be applied to the simultaneous preparation of many chemically distinct compounds. In exchange for the ability to prepare many compounds in a short time frame, compromises are accepted that may result in sacrificing, within acceptable limits, yield and purity of individual reaction products. This entry deals with the basic issues faced in preparing and screening synthetic combinatorial libraries and some of the relevant technologies and methods. No attempt has been made to provide exhaustive coverage of the rapidly expanding range of chemistries now amenable to library synthesis, nor have we attempted to cover the application of combinatorial chemistry as it applies directly to the field of drug discovery.
Combinatorial chemistry seeks to create all possible combinations of a set of building blocks and then extract the identity of members that exhibit a desired property. Thus, if a traditional synthesis involves incorporating building block A, followed by B, and then C to give the desired compound A–B–C, then the analogous combinatorial synthesis would employ a set of monomers, A1–An, followed by a second set of monomers, B1–Bn, and then a third set of monomers, C1–Cn, giving all possible combinations of (A1 to n)–(B1 to n)–(C 1 to n). Such processes can be described as nk sets, where n is the number of building blocks used in each cycle of synthesis and k is the total number of synthetic cycles. The number of individual species generated by these approaches become vast very quickly. For example, a relatively short DNA library of 100 random nucleotide positions has a theoretical complexity of 4100, or 1.6 × 1060 different sequences. This number exceeds the number of protons in the sun. Complexities of more realistic peptide libraries using the 20 amino acids incorporated into proteins are as follows: A library of all possible tripeptides will contain 8000 (203(distinct peptide sequences; a library of all possible tetrapeptides will contain 160,000 (204) distinct peptide sequences; and a library of all possible pentapeptides will contain 3,200,000 (205) distinct peptide sequences. Specialized methods are required to prepare, assay, and elucidate the identity of active compounds from collections of this size. In order to facilitate product purification, compounds are typically synthesized while covalently attached to inert solid supports, including polymeric pins or resin beads, although other materials have been used. Use of solid phase synthesis enables many reactions to be carried out simultaneously using parallel arrays of individual reaction chambers. If each reaction yields a single product, the result is a library of spatially discrete compounds. This approach has the advantage that the identity of compounds can be ascertained immediately, based on the position in the synthetic array and the associated synthetic history. The initial application of this approach was reported by Geysen et al. for synthesis of peptide libraries (1); however, many variations have been reported since then, involving a wide range of chemistries. Although convenient and straightforward to implement, the number of compounds that can be synthesized and screened using this approach is limited by the physical dimensions of the arrays to approximately 10,000 different species. Miniaturization and photolithographic masking techniques have enabled arrays containing more than 100,000 compounds to be synthesized on silica chips (2). Although this has been applied to a variety of chemistries, it is perhaps most widely recognized as the DNA chip technology of Affymetrix (3). Unfortunately, this approach requires highly specialized instrumentation for the photolithographic masking, as well as for assay of the resulting arrays.
The synthesis of larger libraries requires pooling methods that overcome the size limitations of spatial arrays (4). Pooling methods employ mixtures of reactants at each synthetic step (5). For example, in random DNA oligonucleotide synthesis, two or more deoxyribonucleotide phosphoramidites are introduced during each coupling cycle. One major problem with mixture synthesis is that variation in reactivity can result in some building blocks being under- or overrepresented in the final compound pool. At least in some cases, the initial concentration of reactants can be adjusted to correct for these variations in reactivity. A more attractive approach, first introduced by Furka, is the “split synthesis” strategy (6). Briefly, a portion of synthesis resin is “split” or divided into equal portions and placed into individual reaction vessels (Fig. 1). Each vessel receives a single building block in large excess, which drives the reaction to completion. The resin from all the vessels is then recombined, mixed thoroughly, and then redistributed. A second set of building blocks is then introduced. This process is repeated the desired number of cycles, to produce all possible combinations of the desired building blocks. Because each resin bead is exposed to a single building block at each step, there will be only one compound (but many copies) attached to each bead. This has led to the term one-bead, one-compound (OBOC) libraries (7, 8).
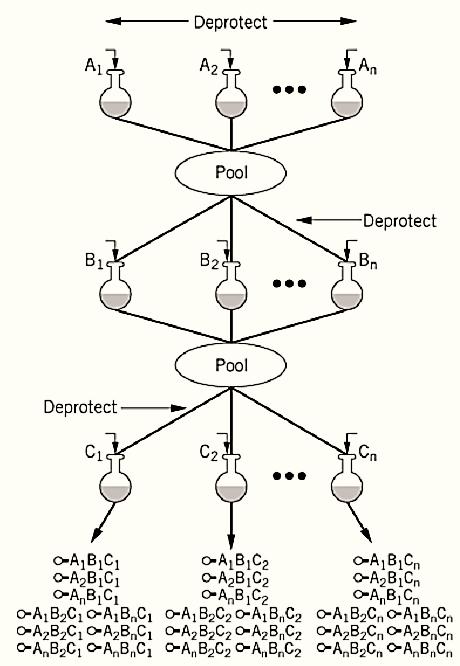
Figure 1. Schematic flow of a split synthesis. The resin is first divided into n equal portions. Following deprotection, each resin batch is reacted with a different monomer from set A. The resin is pooled, mixed, and divided into equal portions. After deprotection, individual monomers from set B are coupled. The process is repeated, and monomers from set C are added. Each final reaction batch is held separately for subsequent assay
Compound identification is straightforward for spatially discrete libraries; however, identifying active species in the large and complex mixtures that result from pooling strategies presents a formidable problem. Typically, each member of a pool is present in a quantity that, although sufficient for determining biological activity, is insufficient for isolation and structure determination by standard analytical methods. As a result, strategies to identify the active members must be built into the synthetic schemes and closely coupled to the types of assays that will be performed with a particular library.
Compounds can be identified from soluble libraries using deconvolution strategies such as those introduced by Houghten (9, 10). This approach relies on cycles of synthesis and screening to focus progressively in on active species. An initial library is prepared in which one position of diversity is held constant. Each individual pool of compounds contains a different building block at the constant position but a mixture of building blocks at the remaining positions of diversity. Screening identifies the pool and therefore the optimal building block at the defined location. A second library is then synthesized in which all members have the optimal building block at this first position, but each pool contains a different building block at a second position of diversity. This process is repeated until the preferred building block for each position is determined. The chief disadvantage of this approach is that it is a laborious and time-consuming process. Moreover, this strategy does not guarantee that the most potent library member will be identified since selection of a “preferred” building block depends on the cumulative activity of a pool. Selection of a particular building block could, therefore, result from the presence in a pool of a relatively few number of potent compounds or a large number of relatively poor inhibitors.
Several strategies have been developed that allow the active members of a library to be determined without requiring cycles of synthesis and screening. The method of positional scanning (11) involves synthesizing a series of combinatorial libraries, each fixing a single position of diversity. Synthesis of each library is divided such that each component pool is prepared using a single building block at the defined position, while the remaining positions are synthesized using mixtures of building
blocks. Although this method does not require resynthesis, it does require additional synthetic effort to prepare a complete set of scanning libraries, and the total number of syntheses is greater than for deconvolution approaches. Moreover, because each position is defined independently, there is an increased chance of missing the most active members in the library.
Orthogonal pooling strategies also avoid the resynthesis burden of deconvolution methods (12). In this approach, two separate libraries are synthesized such that any given pool of the first library has only one compound in common with any pool of the second library. Determining the common species between positive pools from the two libraries reveals the identity of active compounds. The chance for misidentifying the optimal building block at any given position is avoided by this approach, but there is a considerable synthetic burden, as well as compound tracking issues.
The second general assay format is designed to measure the interaction of a target (enzyme, receptor, cell, etc) with compounds while they remain attached to the solid support used for library synthesis. Geysen pioneered this approach for assaying libraries of peptides arrayed on pins (1); however, the full potential was realized only when it was coupled to screening of OBOC libraries (8). Because each resin bead in an OBOC library carries a single library compound and the beads are physically distinct, each bead behaves as an isolated assay system. Briefly, a target is incubated with a pool of beads. The excess target is removed by washing, leaving behind only target that associates specifically with a compound attached to a resin bead. The active compounds are identified following visualization of those beads to which the target specifically interacts.
Specialized techniques are required to identify the compounds attached to individual beads, because each bead can carry at most a few nanomoles of compound, which is insufficient for structure determination except in a few rare cases (eg, peptides). These methods seek to “tag” or “encode” the beads with the synthetic history in a manner that is easily deciphered (13, 14). The code is introduced either prior to library synthesis or following each cycle of building block addition. In the latter case, the chemistries for library synthesis and code synthesis must be mutually compatible, which is one of the major obstacles in the development and utilization of this approach. Examples of encoding methods include the use of peptides read by Edman Degradation (15), nucleic acids read by PCR amplification and sequencing (16), several small molecule chemical classes identified by various analytical methods (17, 18), nonradioactive isotopic tags read by mass spectrometry (19,( and radioisotopes encapsulated within the solid-phase support (20, 21). The development of encoding methods is an active area of research because the split synthesis method is currently the only tractable approach to preparing large libraries in which all possible compounds are represented.
References
1. H. M. Geysen, R. H. Meleon, and S. J. Barteling (1984) Proc. Natl. Acad. Sci. USA 81, 3998–4002.
2. S. P. Fodor, J. L. Read, M. C. Pirrung, L. Stryer, A. T. Lu, and D. Solas (1991) Science 251, 767-773.
3. A. C. Pease, D. Solas, E. J. Sullivan, M. T. Cronin, C. P. Holmes, and S. P. Fodor (1994) Proc. Natl. Acad. Sci. USA 91, 5022–5026.
4. C. Pinilla, J. Appel, S. Blondelle, C. Dooley, B. Dörner, J. Eichler, J. Ostresh, and R. A. Houghten (1995) Biopolymers (Peptide Sci.) 37, 221–240.
5. H. M. Geysen, S. J. Rodda, and T. J. Mason (1986) Mol. Immunol. 23, 709–715.
6. A. Furka, F. Sebestyen, M. Asgedom, and G. Dibo (1991) Int. J. Pept. Protein Res. 37, 487–493.
7. K. S. Lam, M. Lebl, and V. Krchnak (1997) Chem. Rev. 97, 411–448.
8. K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski, and R. J. Knapp (1991(Nature 354, 82–84.
9. C. T. Dooley, N. N. Chung, B. C. Wilkes, P. W. Schiller, J. M. Bidlack, G. W. Pasternak, and R. A. Houghten (1994) Science 266, 2019–2022.
10. S. E. Blondelle, E. Takahashi, P. A. Weber, and R. A. Houghten (1994) Antimicrob. Agents Chemother. 38, 2280–2286.
11. R. A. Houghten, C. Pinilla, S. E. Blondelle, J. R. Appel, C. T. Dooley, and J. H. Cuervo (1991( Nature 354, 84–86.
12. B. Deprez, X. Williard, L. Bourel, H. Coste, F. Hyafil, and A. Tartar (1995) J. Am. Chem. Soc. 117, 5405–5406.
13. S. Brenner and R. A. Lerner (1992) Proc. Natl. Acad. Sci. USA 89, 5381–5383.
14. K. D. Janda (1994) Proc. Natl. Acad. Sci. USA 91, 10779–10785.
15. J. M. Kerr, S. C. Banville, and R. N. Zuckermann (1993) J. Am. Chem. Soc. 115, 2529–2531.
16. M. C. Needels, D. G. Jones, E. H. Tate, G. L. Heinkel, L. M. Kochersperger, W. J. Dower, R. W. Barrett, and M. A. Gallop (1993) Proc. Natl. Acad. Sci. USA 90, 10700–10704.
17. M. H. J. Ohlmeyer, R. N. Swanson, L. W. Dillard, J. C. Reader, G. Asouline, R. Kobayashi, M. Wigler, and W. C. Still (1993) Proc. Natl. Acad. Sci. USA 90, 10922–10926.
18. Z.-J. Ni, D. Maclean, C. P. Holmes, M. M. Murphy, B. Ruhland, J. W. Jacobs, E. M. Gordon, and M. A. Gallop (1996). J. Med. Chem. 39, 1601–1608.
19. H. M. Geysen, C. D. Wagner, W. M. Bodnar, C. J. Markworth, G. J. Parke, F. J. Schoenen, D. S. Wagner, and D. S. Kinder (1996) Chem. Biol. 3, 679–688.
20. K. C. Nicolaou, X.-Y. Xiao, Z. Parandoosh, A. Senyei, and M. P. Novz (1995) Angew. Chem. Int. Ed. English 34, 2289–2291.
21. E. J. Moran, S. Sarshar, J. F. Cargill, M. M. Shahbaz, A. Lio, A. M. M. Mjalli, and R. W. Armstrong (1995) J. Am. Chem. Soc. 117, 10787–10788.

|
|
|
|
تفوقت في الاختبار على الجميع.. فاكهة "خارقة" في عالم التغذية
|
|
|
|
|
|
|
أمين عام أوبك: النفط الخام والغاز الطبيعي "هبة من الله"
|
|
|
|
|
|
|
قسم شؤون المعارف ينظم دورة عن آليات عمل الفهارس الفنية للموسوعات والكتب لملاكاته
|
|
|
















