
آخر المواضيع المضافة

 علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية
علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية | Transition state stabilization |


|
|
Read More
Date: 8-7-2019
Date: 12-5-2017
Date: 31-10-2019
|
One of the most important ways that an enzyme catalyzes any given reaction is through entropy reduction: by bringing order to a disordered situation (remember that entropy is a component of Gibbs Free Energy, and thus a component of the activation energy). Let’s turn again to our previous example (from section 6.1C) of a biochemical nucleophilic substitution reaction, the methylation of adenosine in DNA. The reaction is shown below with non-reactive sections of the molecules depicted by variously shaped 'bubbles' for the sake of simplicity.

In order for this reaction to occur, the two substrates (reactants) must come into contact in precisely the right way. If they are both floating around free in solution, the likelihood of this occurring is very small – the entropy of the system is simply too high. In other words, this reaction takes place very slowly without the help of a catalyst.
Here’s where the enzyme’s active site pocket comes into play. It is lined with various functional groups from the amino acid main and side chains, and has a very specific three-dimensional architecture that has evolved to bind to both of the substrates. If the SAM molecule, for example, diffuses into the active site, it can replace its (favorable) interactions with the surrounding water molecules with (even more favorable) new interactions with the functional groups lining the active site.
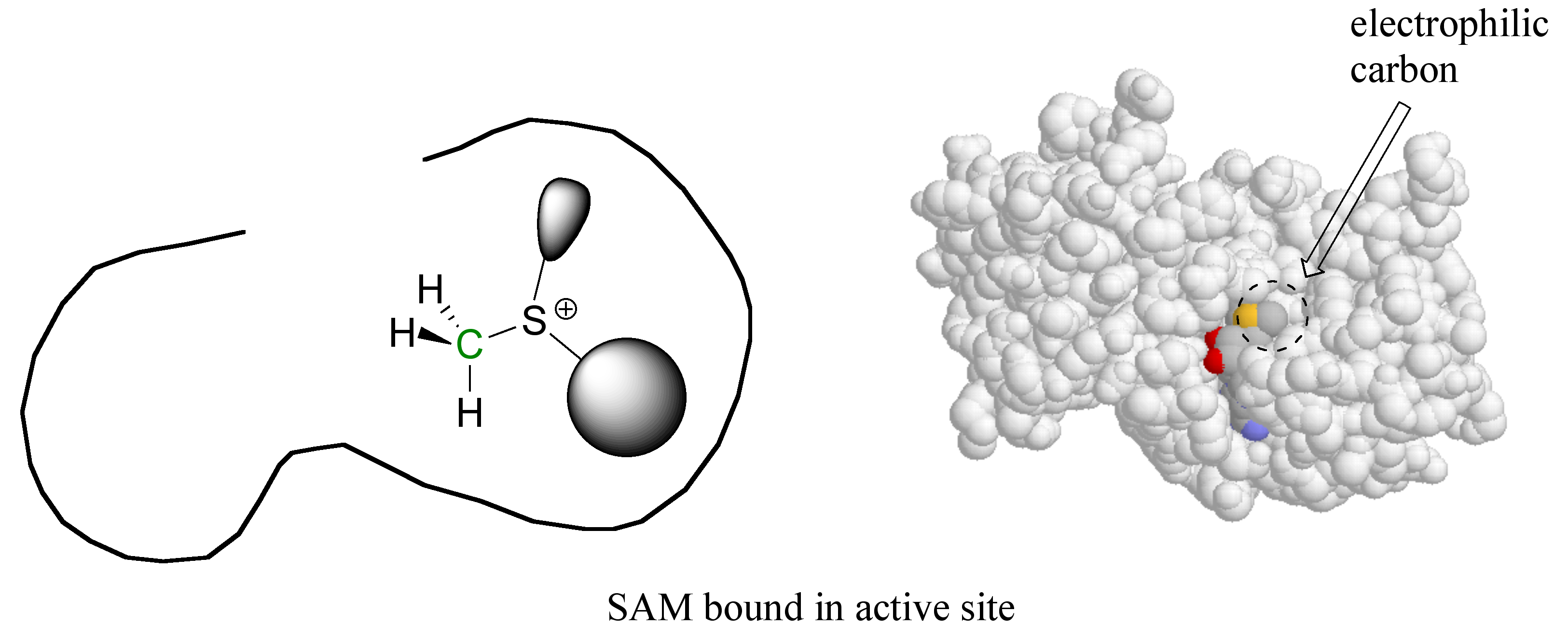
In a sense, SAM is moving from one solvent (water) to another 'solvent' (the active site), where many new energetically favorable interactions are possible. Remember: these new contacts between SAM and the active site groups are highly specific to SAM and SAM alone – no other molecule can ‘fit’ so well in this precise active site environment, and thus no other molecule will be likely to give up its contacts to water and bind to the active site.
The second substrate also has a specific spot reserved in the active site. (Because in this case the second substrate is a small segment of a long DNA molecule, the DNA-binding region of the active site is more of a 'groove' than a 'pocket').
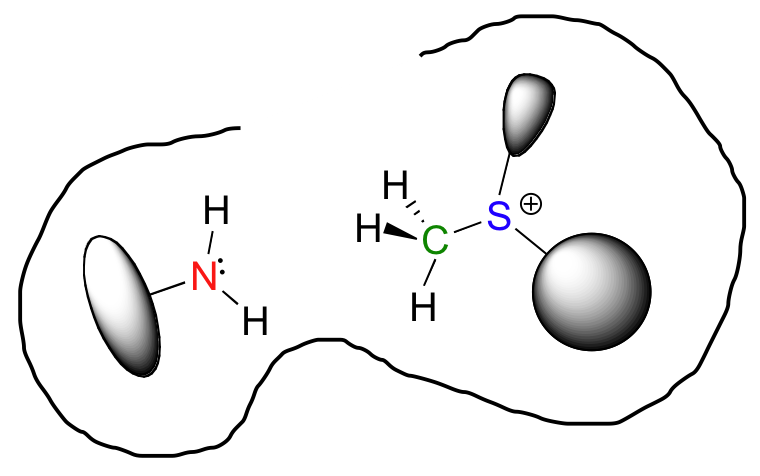
So now we have both substrates bound in the active site. But they are not just bound in any random orientation – they are specifically positioned relative to one another so that the nucleophilic nitrogen is held very close to the electrophilic carbon, with a free path of attack. What used to be a very disordered situation – two reactants diffusing freely in solution – is now a very highly ordered situation, with everything set up for the reaction to proceed. This is what is meant by entropy reduction: the entropic component of the energy barrier has been lowered.
Looking a bit deeper, though, it is not really the noncovalent interaction between enzyme and substrate that are responsible for catalysis. Remember: all catalysts, enzymes included, accelerate reactions by lowering the energy of the transition state. With this in mind, it should make sense that the primary job of an enzyme is to maximize favorable interactions with the transition state, not with the starting substrates. This does not imply that enzyme-substrate interactions are not strong, rather that enzyme-TS interactions are far stronger, often by several orders of magnitude. Think about it this way: if an enzyme were to bind to (and stabilize) its substrate(s) more tightly than it bound to (and stabilized) the transition state, it would actually slow down the reaction, because it would be increasing the energy difference between starting state and transition state. The enzyme has evolved to maximize favorable noncovalent interactions to the transition state: in our example, this is the state in which the nucleophilic nitrogen is already beginning to attack the electrophilic carbon, and the carbon-sulfur bond has already begun to break.
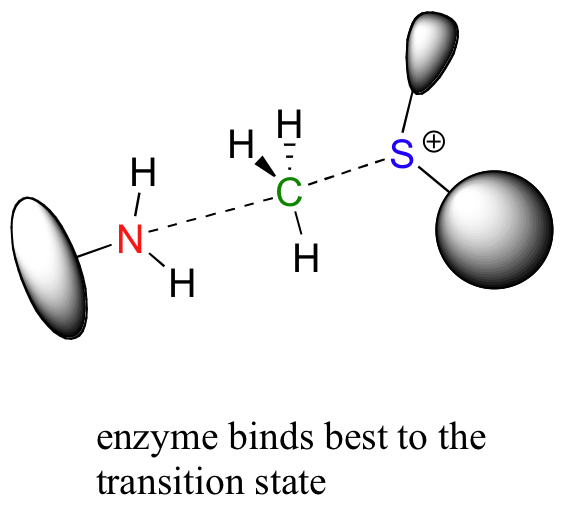
In many enzymatic reactions, certain active site amino acid residues contribute to catalysis by increasing the reactivity of the substrates. Often, the catalytic role is that of acid and/or base. In our DNA methylation example, the nucleophilic nitrogen is deprotonated by a nearby aspartate side chain as it begins its nucleophilic attack on the methyl group of SAM. We will study nucleophilicity in greater detail in chapter 8, but it should make intuitive sense that deprotonating the amine increases the electron density of the nitrogen, making it more nucleophilic. Notice also in the figure below that the main chain carbonyl of an active site proline forms a hydrogen bond with the amine, which also has the effect of increasing the nitrogen's electron density and thus its nucleophilicity (Nucleic Acids Res. 2000, 28, 3950).

How does our picture of enzyme catalysis apply to multi-step reaction mechanisms? Although the two-step nucleophilic substitution reaction between tert-butyl chloride and hydroxide (section 6.1C) is not a biologically relevant process, let’s pretend just for the sake of illustration that there is a hypothetical enzyme that catalyzes this reaction.

The same basic principles apply here: the enzyme binds best to the transition state. But therein lies the problem: there are two transition states! To which TS does the enzyme maximize its contacts?
Recall that the first step – the loss of the chloride leaving group to form the carbocation intermediate – is the slower, rate-limiting step. It is this step that our hypothetical enzyme needs to accelerate if it wants to accelerate the overall reaction, and it is thus the energy of TS1 that needs to be lowered.
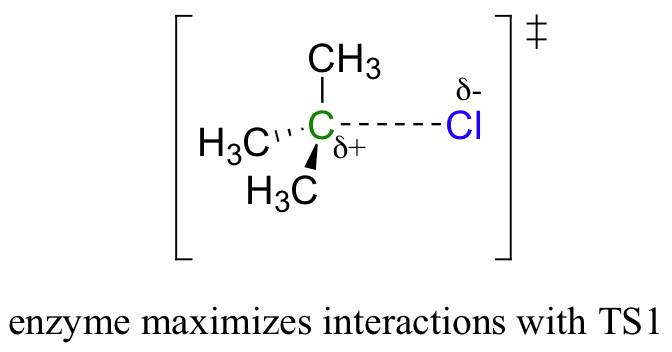
By Hammond’s postulate, we also know that the intermediate I is a close approximation of TS1. So the enzyme, by stabilizing the intermediate, will also stabilize TS1 (as well as TS2) and thereby accelerate the reaction.
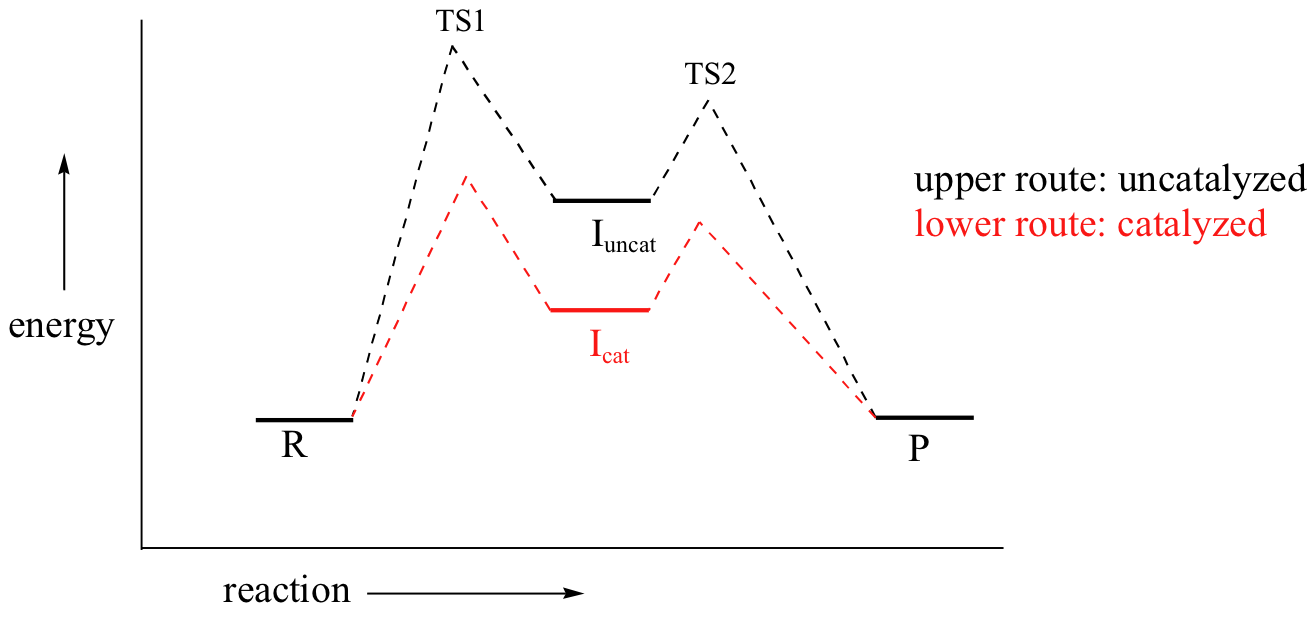
If you read scientific papers about enzyme mechanisms, you will often see researchers discussing how an enzyme stabilizes a reaction intermediate. By virtue of Hammond's postulate, they are, at the same time, talking about how the enzyme lowers the energy of the transition state.
An additional note: although we have in this section been referring to SAM as a 'substrate' of the DNA methylation reaction, it is also often referred to as a coenzyme, or cofactor. These terms are used to describe small (relative to protein and DNA) biological organic molecules that bind specifically in the active site of an enzyme and help the enzyme to do its job. In the case of SAM, the job is methyl group donation. In addition to SAM, we will see many other examples of coenzymes in the coming chapters, a number of which - like ATP (adenosine triphosphate), coenzyme A, thiamine, and flavin - you have probably heard of before. The full structures of some common coenzymes are shown in table 6 in the tables section.


|
|
|
|
تفوقت في الاختبار على الجميع.. فاكهة "خارقة" في عالم التغذية
|
|
|
|
|
|
|
أمين عام أوبك: النفط الخام والغاز الطبيعي "هبة من الله"
|
|
|
|
|
|
|
قسم شؤون المعارف ينظم دورة عن آليات عمل الفهارس الفنية للموسوعات والكتب لملاكاته
|
|
|
















