
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Affinity Chromatography |


|
|
Read More
Date: 28-3-2021
Date: 1-1-2016
Date: 9-11-2020
|
Affinity Chromatography
1. The Basic Principle
Affinity chromatography (AC) is a general chromatographic method for the selective extraction and purification of biological macromolecules on the basis of their biorecognition (1-3). The method makes use of the specific physiological affinity between a desired macromolecule (M) and one of its physiological ligands (L). The ligand, or its analogue (L′), actually acts as a “bait” and is used to extract or “fish out” a desired macromolecule (M1) (Fig. 1) from a mixture of macromolecules (M1; M2; M3; M4; M5; M6¼). The other macromolecules have a very low (if any) affinity for L, presumably because they are designed to refrain from interfering in vivo with the physiological recognition of L by M.
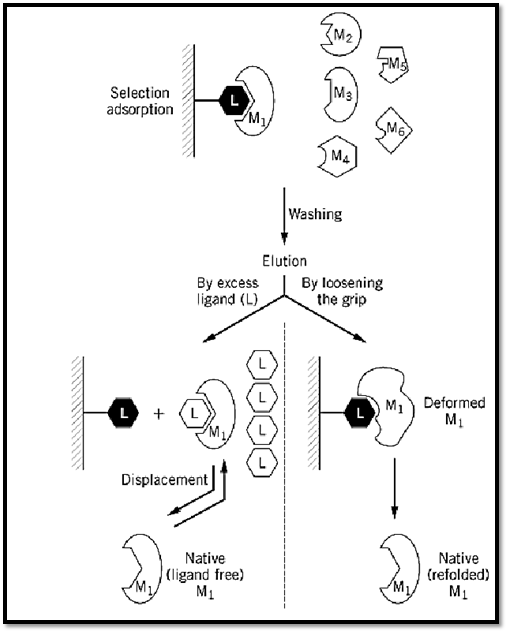
Figure 1. Schematic representation of the general procedure for an AC purification. For further details, see text.
2. General Procedure for an AC Purification: Key Steps
1. Immobilization (anchoring) of the ligand on an inert carrier: L is anchored on a carrier to yield an insoluble material, usually in a beaded form. This carrier should be as inert as possible (eg, beaded agarose) to achieve true active-site-mediated AC. Also, the attachment point of the ligand should not involve groups that are involved in binding to the macromolecule. Over the years, different carriers and various methods for ligand immobilization were developed. These were reviewed and evaluated by Wilchek et al. (3). In general, the anchoring of L onto an inert carrier involves (i) the introduction of chemically reactive groups to the inert carrier, (ii) the covalent attachment of L to the activated carrier, and (iii) inactivation of the excess of reactive groups (if any) that may remain on the activated carrier after completion of the ligand anchoring step. The immobilized ligand can be used either batchwise or as a column. It may also find other uses—for example, to detect or demonstrate specific protein–protein interactions by the binding of a specific protein. Furthermore, it may use the resulting column material to bind and fish out another protein that interacts with it. Such immobilized ligands have been used also for labeling of cells, for the localization of proteins on cell surfaces, for the demonstration of leakage of enzymes or specific proteins from damaged tissues, and so on.
Historically, the pioneering work of Axén et al. (4) on the CNBr activation of beaded agarose had a great influence on the development of AC and the conversion of this methodology into a most widely used tool in separation science. To this day, beaded agarose continues to be the inert carrier of choice, and its activation with CNBr for ligand binding is still an activation method of choice. A thorough analytical study of the mechanism of activation of agarose by CNBr (5) showed that three major products are formed: a carbamate (chemically inert), a linear or a cyclic imidocarbonate (slightly reactive), and a cyanate ester (chemically very reactive). Analysis of freshly activated agarose showed that 60% to 85% of the total coupling capacity of the agarose is due to the formation of the cyanate esters. They are the ones that actually react and immobilize the ligand (Fig. 2). On the basis of this mechanism of activation by CNBr, it became possible to develop more efficient activation procedures, which are reviewed in Refs. 3 and 6.
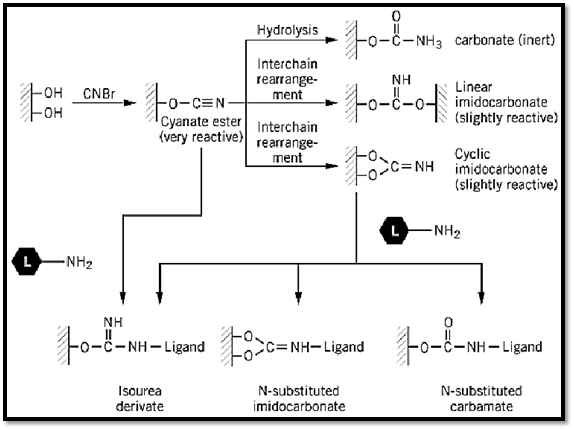
Figure 2. The mechanism of the CNBr activation of agarose. (Modified from Ref. 3.)
2. Selective adsorption. The key selective step in AC is obviously the extraction of the desired macromolecule M, which is singled out and removed by the immobilized L, from the mixture in which it is present. The macromolecule—be it an enzyme, an antibody, a receptor, a hormone, a growth factor, or the like—is selectively bound by the biospecific ligand L, which can be another protein, a peptide, a polynucleotide or a nucleotide, a polysacharide or a carbohydrate, a lipid, a vitamin, or just a metal ion. Functionally, L may be a substrate, a substrate analogue, an inhibitor, an antigen, a coenzyme, a cofactor, or a regulatory metabolite. In many cases, the biospecific ligand used for the immobilization is a structural analogue of the physiological ligand (L′). It is imperative, however, to ensure that it still retains the property of selective binding to M, and ideally to M only. In choosing the ligand for an affinity chromatography column, it is often possible to adjust the grip of M onto the anchored L, and thus to optimize both the adsorption and the elution steps. It should be noted that the adsorption conditions used (buffer, pH, ionic strength, temperature) should also be carefully chosen to secure an optimal and selective adsorption.
3. Washing out nonspecifically bound impurities. This is usually carried out with an excess of the buffer used for selective adsorption.
4. Elution of the desired macromolecule. The detachment of M from the column (elution) is one of the most important steps in purification by AC. Obviously, the ideal elution is by a specific displacement of M with an excess of its biospecific ligand (Fig. 1). This procedure preserves the native structure of M by forming the more stable complex of M with its biospecific ligand L. When such elution is achieved, it strongly suggests that true active-site-mediated AC is involved. However, very often biospecific ligands fail to elute the desired protein, and nonspecific means have to be applied. These usually include a change in solvent or buffer composition, a change in pH or in ionic strength, the addition of a chaotropic or a “deforming” buffer, a change in temperature, or a change in the electric field (electrophoretic desorption) (3). All these bring about a deformation of the protein (7, 8), a concomitant loosening of the grip of M for L, and consequently elution. In some cases, the binding of M to the L column is so tight that it is not possible to recover M in a fully active form. If M is an enzyme, this may yield a less active preparation (part of the M molecules may be totally inactive, or all molecules may have a lower affinity for the substrate or a lower turnover number). In some instances, the purified enzyme is fully active, but it may lose its ability to be regulated—for example, if the regulatory domain of M loses its affinity for a regulatory metabolite.
Under such circumstances, immobilized ligands with lower affinity for M must be tried. Among the remedies that can be used to improve the elution step, one should note the possibility of binding the ligand to the matrix by means of an easily cleavable form—for example, through an ester bond (9,( 10, which can be readily hydrolyzed with a mild base; through a link that includes vicinal hydroxyl groups, which can be readily cleaved with periodate; or through diazo bonds, which can be readily reduced with dithionates (11). It should be remembered, however, that such columns are of limited value, because they can be used only once. Electrophoresis has also been used for elution (12). Because proteins are charged, they will detach from the column and migrate toward the appropriate electrode, if the column with the adsorbed M is exposed to a strong enough electric field. This mild method of elution was successfully applied with high yields in immunoaffinity chromatography and in some AC systems.
3. Interposing an “Arm” Between the Ligand and the Matrix Backbone
While developing the basic principles of AC, it was observed that the purification of M is often improved by interposing a hydrocarbon chain (an “arm” or a “spacer”) between L and the matrix backbone (1). It was presumed that such an arm relieves the steric restrictions imposed by the backbone on the ligand, thereby increasing its flexibility and its availability to the protein (13). Such arms were found to improve significantly the extraction of proteins and the efficacy of the purification by AC. Initially, it was assumed that such hydrocarbon arms do not alter the inert nature of the matrix, a condition that obviously has to be ensured to preserve an active-site mediated adsorption of the extracted protein. This assumption seemed reasonable at the time because it had just been shown that at least some water-soluble proteins are quite well described as “an oil drop with a polar coat” (14), implying that the surface of water-soluble proteins is polar and thus not attracted to lipophilic “baits.” We now know that such arms, in and of themselves, may bind proteins. In fact, this observation led to the discovery of hydrophobic chromatography.
4. The Limitations of Biospecificity—Interactions that are not Active Site-Mediated
Proteins and their physiological ligands are multifunctional molecules whose functions involve a variety of physical interactions: hydrophobic, electrostatic, ion-dipole, and so on. Therefore, it is reasonable to assume that a protein might interact with a column coated with a ligand (very often anchored to the beads at a local concentration much higher than its concentration in vivo) not only by means of its active site. While it is sometimes possible to minimize these nonspecific interactions, it is not always possible to avoid such interfering effects, because they may be an intrinsic property of the system. For example, if ATP is linked to a matrix through its amino group or its ribose moiety; the column thus obtained may retain an enzyme having a biospecific site for ATP; but at the same
time, this very column would be negatively charged due to its triphosphate groups, and it would have hydrophobic loci due to its adenine residues. Other proteins, in addition to the desired one, may therefore “regard” the column material as an ion exchanger by virtue of its triphosphate groups, or as a hydrophobic column by virtue of its adenine moieties. The efficiency of resolution will then depend on the magnitude of the affinity produced by charge–charge or hydrophobic interactions, as compared to the affinity between the active site of the desired macromolecule and its immobilized substrate or effector analogue. With columns of macromolecular ligands (eg, enzyme subunits, antibodies, lectins), the probability of encountering such built-in interfering effects is considerably higher, because their immobilization usually involves different anchoring points. This leads to a heterogeneous presentation of the various regions of the ligand macromolecule. In some of these presentations, the biospecific active site is available for interaction, while in other presentations the active site itself is inaccessible or sterically hindered. Hydrophobic patches in such ligands may be available for interaction not only in the biospecifically functional presentation, but also in other presentations. In fact, the tendency of a lectin such as concanavalin A to adsorb onto hydrophobic substances, in addition to its binding to sugars of the mannosyl configuration, was observed in several laboratories.
5. The Relativity of Biological Recognition: Different Proteins may Share a Taste for a Biorecognition Elements
The occurrence of common biorecognition sites in different enzymes is obvious when they are functionally similar, acting on the same substrate (eg, ATP), or utilizing the same cofactor (eg, NAD). This actually forms the basis for general ligand-affinity chromatography (15). However, common biorecognition elements may also be found with proteins having no apparent functional similarity. For example, the free catalytic subunit of cAMP-dependent protein kinase (protein kinase A) is preferentially retarded on immobilized soybean trypsin inhibitor (16). Though initially unexpected, this is actually not surprising; in spite of the fact that trypsin and this kinase catalyze two different chemical reactions (hydrolysis of peptide bonds versus a phosphotransferase reaction), these two enzymes do have similar biorecognition elements (or subsites) at their active site: trypsin cleaves peptide bonds adjacent to positively charged amino acid residues (arginine and lysine), while cAMP-dependent protein kinase phosphorylates serine residues that are vicinal (in the sequence of amino acids) to the same positively charged arginine and lysine residues (17-20). Similarly, it was shown (21) that TLCK (a-N-tosyl-L-lysine chloromethyl ketone), an affinity labeling reagent originally designed for labeling the active site of trypsin, specifically attacks a thiol group at the active site of the catalytic subunit of cAMP-dependent protein kinase. It seems, therefore, that the retardation of the free catalytic subunit on the immobilized inhibitor is due (at least in part) to an affinity between the inhibitor and recognition subsites at the active site of the enzyme.
References
1. P. Cuatrecasas, M. Wilchek, and C. B. Anfinsen (1968) Proc. Natl. Acad. Sci. USA 61, 636.
2. P. Cuatrecasas and C. B. Anfinsen (1971) Annu. Rev. Biochem. 40, 259.
3. M. Wilchek, T. Miron, and J. Kohn (1984) Methods Enzymol. 104, 3.
4. R. Axén, J. Porath, and S. Ernbäck (1967) Nature (London) 214, 1302.
5. J. Kohn and M. Wilchek (1981) Anal. Biochem. 115, 375.
6. S. B. Mohan and A. Lyddiatt (1997) In Affinity Separations (P. Matejtschuk, ed.), IRL Press, Oxford University Press, New York, p. 1.
7. S. Shaltiel, J. L. Hedrick, and E. H. Fischer (1966) Biochemistry 5, 2108.
8. J. L. Hedrick, S. Shaltiel, and E. H. Fischer (1969) Biochemistry 8, 2422.
9. R. J. Brown, N. E. Swaisgood, and H. R. Horton (1979) Biochemistry 18, 4901.
10. P. Singh, S. D. Lewis, and J. A. Shafer (1979) Arch. Biochem. Biophys. 193, 284.
11. P. Singh, S. D. Lewis, and J. A. Shafer (1980) Arch. Biochem. Biophys. 203, 776.
12. M. R. Morgan, P. J. Brown, M. J. Lieiand, and P. D. Ocan (1978) FEBS Lett. 87, 239.
13. P. Cuatrecasas (1970) J. Biol. Chem. 245, 3059.
14. D. C. Phillips (1966) Sci. Am. November, 78.
15. K. Mosbach (1978) Adv. Enzymol. 46, 205.
16. E. Alhanaty, N. Bashan, S. Moses, and S. Shaltiel (1979) Eur. J. Biochem. 101, 283.
17. H. G. Nimmo and P. Cohen (1977) Adv. Cyclic Nucl. Res. 8, 145.
18. O. Zetterquist et al. (1976) Biochem. Biophys. Res. Comun. 70, 696.
19. B. E. Kemp, E. Benjamin, and E. G. Krebs (1976) Proc. Natl. Acad. Sci. USA 73, 1038.
20. P. Daile, P. R. Carnegie, and J. D. Young (1975) Nature 257, 416.
21. A. Kupfer, V. Gani, J. S. Jimenez, and S. Shaltiel (1979) Proc. Natl. Acad. Sci. USA 76, 3073.

|
|
|
|
دخلت غرفة فنسيت ماذا تريد من داخلها.. خبير يفسر الحالة
|
|
|
|
|
|
|
ثورة طبية.. ابتكار أصغر جهاز لتنظيم ضربات القلب في العالم
|
|
|
|
|
|
|
قسم شؤون المعارف ووفد من جامعة البصرة يبحثان سبل تعزيز التعاون المشترك
|
|
|
















