
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Recombinant Dna Technology and Its Applications |


|
|
Read More
Date: 10-11-2015
Date: 12-11-2015
Date: 11-11-2015
|
Recombinant Dna Technology and Its Applications
INTRODUCTION
Recombinant DNA technology involves techniques of uniting two heterologous DNA molecules using in vitro ligation. The desired fragment of specific DNA sequence within a complex DNA population is selectively amplified using either cell based DNA cloning or polymerase mediated cloning using the polymerase chain reaction (PCR). Cell based DNA cloning involves attaching foreign DNA fragments (target DNA) to DNA sequences capable of independent ligation called vectors or replicons. This is done using an enzyme called DNA ligase, and the process is called ligation. Cutting the target DNA and the vector, with specific restriction endonucleases facilitate this step (Fig. 1A). Following ligation, the next step is called transformation, where the recombinant DNA molecules are transferred into host cells in which they can undergo DNA replication independent of host cell chromosomes. Recombinant screening and identification of cells containing recombinant DNA (vector molecules with inserts) is accomplished by insertional activation of a marker gene. The vector molecule is designed to have a multiple cloning site called the polylinker within the marker gene.
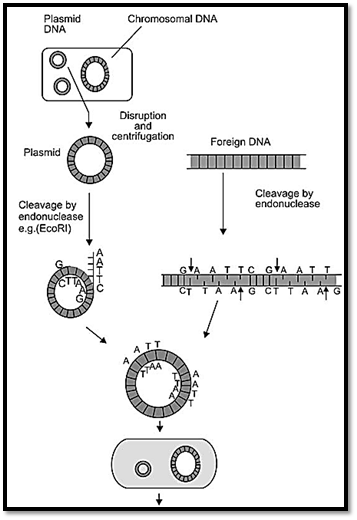
Fig. 1A: Steps in recombinant DNA technology generation of a recombinant plasmid by ECORI
TOOLS OF RECOMBINANT DNA ANALYSIS
Restriction Endonucleases
Restriction endonucleases are enzymes, which cleave DNA at specific recognition sequences, usually 4-8 base pairs long. A DNA sequence that is recognized by a restriction enzyme is called a restriction site. Restriction endonucleases enable the target DNA to be cut up into pieces and facilitate ligation into similarly cut vector molecules. The recognition sequences for a vast majority of restriction endonucleases are palindromes. A palindrome is a DNA sequence that reads the same when read in the 5' to 3' direction on each strand. Restriction fragments generated after cleavage with restriction endonucleases can be blunt ended or possess 5' or 3' overhangs called sticky ends. Restriction endonucleases that happen to recognize the same target sequence are called isoschizomers. A restriction enzyme is named according to the organism from which it was isolated. The first letter of the name is from the genus of the bacteria, the next two letters are from the name of the species, an additional subscript letter indicates the type of strain and the final number is the order in which the enzyme was discovered in the particular organism. Some examples of restriction endonucleases, their source, and recognition sequence are given below.
AluI is derived from Arthrobacter luteus and the recognition sequence is AGCT; Taq I is derived from Thermus aquaticus and the recognition sequence is TCGA; HindIII is derived from Hemophilus influenzae Rd and the recognition sequence is AAGCTT; EcoRI is derived from Eschericia Coli R factor and the recognition sequence is GAATTC; BamHI is derived from Bacillus amyloliqueficans H and the recognition sequence is GGATTC; SmaI is derived from Serratia marcescens and the recognition sequence is CCCGGG; and NotI is derived from Nocardia otitidis caviarium and the recognition sequence is GCGGCCGC.
VECTOR SYSTEMS
A vector is a molecule of DNA to which the fragment of DNA to be cloned is attached. The vector should be capable of autonomous replication, it must contain specific nucleotide sequences recognized by restriction endonucleases, and it must carry a gene that confers the ability to select for the vector such as an antibiotic resistance gene. Cloning vectors that can accept large DNA inserts have been used in general physical mapping of genomes and have permitted the characterization and expression of large genes or gene complexes. Figure .1B some of the commonly used vectors are described below.
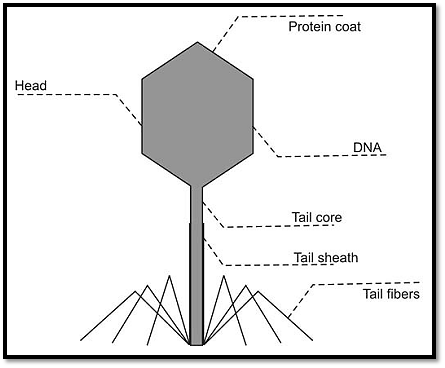
Fig. 1B: Components of bacteriophage vector
Plasmids
Bacteria contain single large circular chromosomes. In addition, most species also contain small circular extra chromosomal double stranded DNA molecules called plasmids, which individually contain very few genes. Their existence is intracellular and they are vertically distributed to daughter cells following host division, or they can be transferred horizontally to neighbouring cells during bacterial conjugation. Plasmid DNA undergoes replication that may or may not be synchronised to chromosomal division. Plasmids may carry genes that convey antibiotic resistance to the host bacterium and may facilitate the transfer of genetic information from one host to the other. If a DNA fragment is inserted into the middle of such an antibiotic resistance gene, then cells carrying the recombinant plasmid will be sensitive to this antibiotic. Thus the pattern of antibiotic resistance can be used to select for and identify bacterial cells carrying recombinant plasmids. Another method is using fi-galacatosidase gene complementation. Plasmids can be readily isolated from bacterial cells, their circular DNA cleaved at specific sites by restriction endonucleases, and foreign DNA inserted into them. The hybrid plasmid can be reintroduced into a bacterium and large numbers of copies of plasmid containing the foreign DNA can be produced. Foreign DNA molecules 0-10 kb in size can be cloned using such vectors.
Bacteriophage λ/Phage Vectors
Phages, also known as bacteriophage λ are viruses, which infect bacteria, and are 45 kb in size. DNA is cloned in and the chimeric DNA is collected after the phage proceeds through its lytic cycle and produces mature infective phage particles. In order to design suitable cloning vectors based on λ, foreign DNA needs to be attached to the λ replicon in vitro, and the resultant recombinant DNA be able to be transformed into E. coli cells at a high efficiency. DNA is packaged in a protein coat resulting in high infection efficiency. Modification of theλresults in two types of vectors. One is the replacement λ vector, which lacks the central segment of the λ genome, which can be replaced by a foreign DNA fragment. These vectors can be used to clone DNA fragments up to 23 kb in length, and such vectors are used to make DNA libraries. The other type is the insertion λ vector, where the λ genome is modified to permit insertional cloning into the cI gene. These vectors are used to make cDNA libraries and can be used to clone fragments up to 10 kb in length.
Cosmid Vectors
Cosmid vectors contain cos sequences inserted into a small plasmid vector. Cos sites are required for packaging λ DNA into the phage particle. Foreign DNA molecules 30-44 kb in size can be cloned using such vectors.
BAC Vectors
Bacterial artificial chromosomes (BACs) contain a low copy number replicon and only very low yields of recombinant DNA can be recovered from host cells. An example is the E. coli fertility plasmid, the F factor. The plasmid contains two genes parA and parB, which makes the copy number of the F factor at 1-2 per E. coli cell. Vectors based on the F factor system are able to accept large foreign DNA fragments greater than 300 kb. The resulting recombinants can be transferred with efficiency into bacterial cells using electroporation, resulting in BACs.
Bacteriophage P1 vectors and PACs
Bacteriophages have relatively large genomes, which allow development of vectors that can accommodate large foreign
DNA fragments. An example is the bacteriophage P1 that packages its genome in a protein coat. P1 cloning vectors are designed in which components of P1 are included in a circular plasmid and can accept up to 100 kb of foreign DNA. The features of the P1 and F factor systems have been combined to produce P1 derived artificial chromosome (PAC) cloning systems. Foreign DNA molecules up to 150 kb in size can be cloned using PACs.
YACs
Cloning of very large fragments involves the construction of yeast artificial chromosomes (YACs) due to the finding that the great bulk of DNA in the chromosome is not required for normal chromosome function. The DNA segment necessary for functional activity in vivo in yeast is limited to a few hundred base pairs of DNA. As a result a novel cloning system was generated based on the use of ARS (autonomous replicating sequence) elements, which are elements required for autonomous replication of chromosomal DNA. To make a YAC, two telomeres, one centromere and one ARS element along with an up to 2 Mb suitably sized foreign DNA fragment is used. The overall transformation efficiency for YACs is very low and so is the yield of cloned DNA (up to one copy per cell). Foreign DNA molecules 0.2 to 2 Mb in size can be cloned into YACs.
GENOMIC AND cDNA LIBRARIES
DNA libraries are comprehensive collections of DNA clones (cloned restriction fragments) from complex starting DNA populations. There are two types of libraries, genomic DNA libraries and cDNA libraries (Fig. 2).
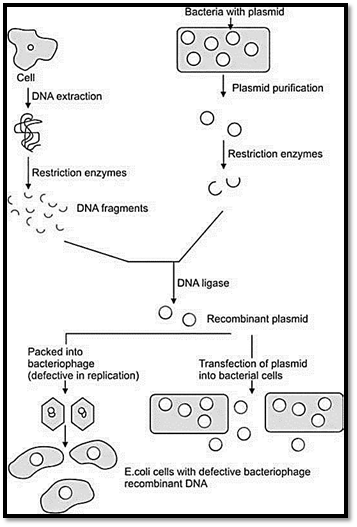
Fig. 2 : Genomic and cDNA libraries
Genomic DNA Libraries
Genomic libraries are collections of fragments of double stranded DNA obtained by digestion of total DNA of an organism with a restriction endonuclease, and subsequent ligation into an appropriate vector. The recombinant DNA molecules are replicated within host bacteria. The amplified DNA fragments represent the entire genome of the organism and are called a genomic library. The complexity or number of independent DNA clones of a genomic DNA library can be defined in term of genome equivalents (GE). A genome equivalent of one, which is a so-called one fold library is obtained when the number of independent clones is equal to genome size/average insert size.
cDNA Libraries
The enzyme reverse transcriptase (RNA dependant DNA polymerase) can be used to make a DNA that is complementary in base sequence to the mRNA called cDNA (complementary cDNA) If a gene of interest is expressed at a very high level in a particular tissue, the mRNA corresponding to that gene is also likely present at high concentrations in the cell. The starting material for making cDNA libraries is total RNA from a specific tissue or specific developmental stage of embryogenesis. The mRNA is used as a template to make a cDNA library using reverse transcriptase and the cDNA can be amplified by cloning or PCR. These mixtures of heterogeneous cDNAs can be cloned to make a cDNA library. To assist cloning, oligonucleotide linkers which contain suitable restriction sites are ligated to each end of the cDNA.
Nucleic acid hybridisation
Nucleic acid hybridisation is a method for identifying closely related nucleic acid molecules within two populations. One is the target of a complex, heterogeneous population of nucleic acid molecules, such as total genomic DNA or RNA. The other, called a probe, is a homogenous population of cloned DNA or chemically synthesized oligonucleotides. The rationale of hybridisation is to use the probe to identify related fragments from the complex target molecules and anneal to them. There are two types of hybridisation assays, standard and reverse assays. Standard nucleic acid hybridisation assays consist of the labelled probe in solution, and the unlabelled target bound to a solid support. Reverse nucleic acid hybridisation assays consist of the labelled target (complex DNA in solution) and unlabelled probes such as oligonucleotides or DNA clones bound to solid support. Examples of standard nucleic acid hybridisation assays include Southern blotting, Northern blotting, and dot blots using allele specific oligonucleotides (ASOs). Examples of reverse nucleic acid hybridisation assays include reverse dot blots, DNA microarrays, and oligonucleotide microarrays. Some examples of these assays are described below.
Dot Blot Hybridisation Assay
Dot blot assay is a screening method in which an aqueous solution of target DNA like total human genomic DNA is spotted onto a nitrocellulose or nylon membrane and allowed to dry. The target sequence is heat or alkali denatured and is exposed to a solution containing single stranded labelled probe. The probe target heteroduplex is allowed to form and the membrane is washed to remove excess non-specific probe, dried and exposed to autoradiographic film. This method employs specific oligonucleotides probes (ASOs) to discriminate between alleles differing at a single nucleotide position. ASO dot blot hybridisation is used to identify common mutations in sickle cell anaemia, and other commonly seen mutations.
Southern Blot Hybridisation Assay
Target DNA is digested with restriction endonucleases, size fractionated by agarose gel electrophoresis, denatured and transferred to a nitrocellulose or nylon membrane for hybridisation. The immobilized single stranded target DNA sequences are allowed to associate with labelled single stranded probe DNA. The radiolabel led probe binds only to complementary sequences in target DNA and can be detected by exposure to autoradiographic film (Fig. 3).
Fig. 3 : Shows a diagrammatic representation and an example of a Southern blot assay
Northern Blot Hybridisation Assay
Northern blotting is a variant of Southern blotting in which the target nucleic acid is RNA instead of DNA. This method is used to obtain information on the expression patterns of specific genes. RNA isolated from a variety of tissues can be run in different lanes and size fractionated. This can be transferred to a membrane and hybridisation carried out with a suitable labelled nucleic acid probe. The data obtained can provide information on the range of tissues in which the gene is expressed and the abundance of transcripts. Different sizes of transcripts are produced due to alternative splicing and can be detected on a Northern blot. Figure 4 shows an example of the use of labelled factor IX cDNA probe, and levels of expression detected in lanes in the upper panel. The lower panel shows hybridisation of the same blot with a universally expressed GAPDH probe showing equal expression in all lanes, which also confirms equal loading of RNA in all lanes.

Fig. 4 : An example of the use of labelled factor IX cDNA probe, and levels of expression detected
In situ Hybridisation
In situ hybridisation involves hybridisation of a nucleic acid probe to the denatured DNA of a chromosome preparation, and example of which is fluorescent in situ hybridisation (FISH) described elsewhere. Nucleic acid probes (double stranded cDNAs or single stranded RNA probes called riboprobes, labelled isotopically or non-isotopically) can also be hybridised to RNAs of tissue sections fixed onto slides called tissue in situ hybridisation or whole organs or embryos called whole mount in situ hybridisation.
Microarray Hybridisation Assay
DNA micro array technologies (DNA chips) employ a reverse nucleic acid hybridisation approach. The probes consist of unlabelled DNA fixed to a solid support (oligonucleotide or DNA arrays) and the target is labelled in solution. Micro arrays of DNA clones are generated by micro spotting, and micro arrays of oligonucleotides are generated by combining photolithography and in situ synthesis of oligonucleotides. The applications of micro array technology include large scale screening of gene expression at the RNA level and screening of DNA variation, including assaying for known mutations in genes and identification of single nucleotide polymorphisms (SNPs).
Western Blotting
This method is used to detect protein expression using cell extracts fractionated according to size using a form of polyacrylamide gel electrophoresis using SDS-PAGE and transfer (blotting) to a membrane. The proteins are detected using antibodies specific to regions of the protein such as specific domains or C or N terminal domains. Antibodies to human gene products are obtained by injecting suitable animals with immunogens such as synthetic peptides or fusion proteins, or by using genetic engineering methods such as phage display technology.
Polymerase chain Reaction
The polymerase chain reaction (PCR) is a rapid in vitro method for amplifying defined target sequences present within a source of DNA. The method is designed to permit selective amplification of a specific target DNA sequence within a heterogeneous collection of DNA sequences like total genomic DNA or a complex cDNA population (Fig. 5).
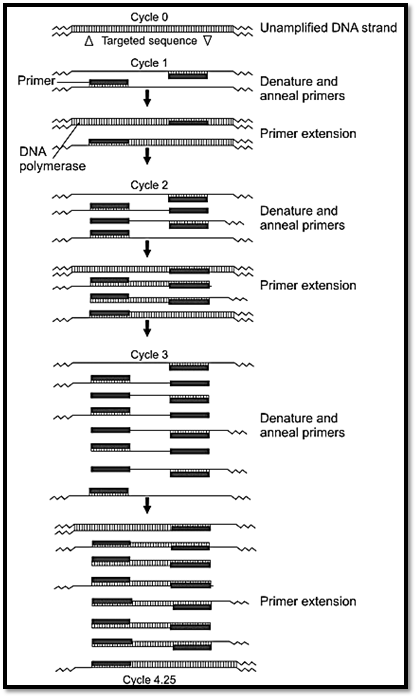
Fig. 5: Polymerase chain reaction (PCR)
Basic Features of PCR
PCR uses a DNA polymerase to repetitively amplify targeted portions of DNA. Each cycle of amplification doubles the amount of DNA in the sample leading to an exponential increase in DNA with repeated cycles of amplification. In order to perform a PCR it is necessary to know the nucleotide sequence of short sequences flanking the region of interest to be amplified. The nucleotide sequences of the flanking regions are used to design and construct two single stranded oligonucleotides, usually 20-30 nucleotides long that are complementary to the respective flanking sequences. These synthetic oligonucleotides are called primers.
There are three steps in a PCR reaction, denaturation, annealing and extension. For denaturation, the DNA to be amplified is heated to separate the double stranded target DNA into single strands. This involves heating the PCR mixture to 93oC for human genomic DNA template. Annealing of primers to single stranded DNA occurs when the temperature is lowered to the temperature that is approximately five degrees below the melting point (Tm) of the primers used in the reaction. For the extension reaction, DNA polymerase and an excess of deoxyribonucleoside triphosphates (dATP, dGTP dCTP dTTP) are added to the mixture to initiate the synthesis of two new chains complementary to the original DNA. This is done at 70oC. DNA polymerase adds nucleotides to the 3’ hydroxyl end of the primer, and strand growth extends across the target DNA making complementary copies of the target. At the completion of one cycle of replication, the reaction mixture is heated again to denature the DNA strands, both the original target strand and the newly generated strands. Each strand binds a complementary primer and the cycle of chain extension is repeated. Typically 20-30 cycles are run during DNA amplification. Each newly synthesised polynucleotide can act as a template for successive cycles, which leads to an exponential increase in the amount of target DNA with each cycle. After about 25 cycles of DNA synthesis the products of the PCR will include in addition to the starting DNA about 105 copies of specific target sequence. This amount is easily visualised as a discrete band of specific size when subjected to agarose gel electrophoresis.
The major advantages of PCR are its rapidity, sensitivity, and robustness. The major disadvantages of PCR are the general requirement for prior target sequence information, the size of the DNA fragments generated and the limited amount of PCR product that is obtained. Another disadvantage is the infidelity of Taq polymerase, which has no associated 3’ to 5’ exonuclease activity to confer a proofreading function, which means the error rate due to misincorporation during DNA replication is high. This can be overcome by using other polymerases such as Pfu polymerase.
Applications of PCR
1- PCR enables rapid amplification of numerous DNA templates for screening of uncharacterised mutations. The identification of exon-intron boundaries and sequencing at the end of introns of a gene of interest offers the possibility of genomic mutation screening by amplification of individual exons by PCR and screening by various mutation screening methods such as single stranded conformational polymorphism analysis (SSCP), heteroduplex analysis, or chemical cleavage mismatch analysis. PCR can also be used to provide amplification of cDNA sequences for mutation screening. To do this mRNA is isolated and converted to cDNA using reverse transcriptase, and the cDNA is used as a template for a PCR reaction. This is called reverse transcriptase PCR or RT-PCR.
2- PCR can be used for rapid typing of polymorphic genetic markers such as RFLPs (restriction fragment length polymorphisms) and STRPs (short tandem repeat polymorphisms).
RFLPs result in alleles possessing or lacking a specific restriction site. Such polymorphisms can be detected using Southern blotting. RFLPs are genetic variants that examined by cleaving DNA into fragments (restriction fragments) with a restriction enzyme. The length of the restriction fragment is altered if the genetic variant alters the DNA to create or abolish a restriction site. Mutation of one or more nucleotides at a restriction site can render the site unrecognisable by the enzyme or create a new restriction site. Cleavage with the enzyme will result in fragments of lengths differing from normal that can be detected by DNA hybridisation. PCR can be used to type RFLPs by designing primers that flank polymorphic restriction sites, amplifying from genomic DNA, and cutting the PCR product with appropriate restriction enzymes and separating the fragments by agarose gel electrophoresis. STRPs are also called microsatellite markers and consist of short sequences that are tandemly repeated several times. An example of these is dinucleotide repeats such as CA repeats, trinucleotide and tetranucleotide repeats. Primers are designed from sequences known to flank a specific STRP locus, permitting amplification of alleles whose sizes differ by integral repeat units. The PCR products can be size fractionated by polyacrylamide gel electrophoresis. An example of the use of a CA repeat marker in an autosomal dominant pedigree is shown in Figure 6.

Fig. 6 : An autoradiograph of a polymorphic tetranucleotide repeat co-segregating in a family with a dominant disorder
3- Use of PCR in genomic DNA cloning and cDNA cloning. Cloning of new members of a DNA family or cloning of cDNAs from amino acid sequence can be carried out by using DOP-PCR. DOP-PCR (degenerate oligonucleotide PCR) is a form of PCR using partially degenerate oligonucleotides to permit searching of a new or uncharacterised DNA sequence that belongs to a family of related sequences either within or between species.
4- PCR can be used for gene expression studies using RT- PCR. Spatial patterns of expression are provided efficiently by tissue in situ hybridisation. Quantitation of expression of a particular gene can also be provided by a Northern blot, which requires large amounts of starting material in the form of RNA. RT-PCR provides a rough quantitation of expression of a particular gene using very small amounts of starting material. RT-PCR can also be useful for identifying and studying different isoforms of an RNA transcript produced due to alternative splicing.
DNA Sequencing
DNA sequencing involves enzymatic DNA synthesis in the presence of base specific dideoxynucleotide chain terminators. Prior to these methods, chemical DNA sequencing methods were employed using base specific chemical modification and subsequent cleavage of DNA. Current methods of DNA sequencing use enzymatic methods. The DNA to be sequenced is provided in a single stranded form, from which DNA polymerase synthesises new complementary DNA strands. The subsequent DNA sequencing reactions involve DNA synthesis using one or more labelled nucleotides and a sequencing primer. In addition to the normal nucleotide precursors, DNA synthesis is carried out in the presence of base specific dideoxynucleotides (ddNTPs). The principle of dideoxy sequencing is that the sequencing primer binds specifically to a region 3’ of the desired DNA sequence and primes synthesis of a complementary DNA strand in the indicated direction. Four base specific reactions are carried out in parallel each with all four dNTPs and one ddNTP. Competition for incorporation into the growing DNA chain between a ddNTP and its normal dNTP analogue results in a population of fragments of different lengths. The fragments have a common 5’ end defined by the sequencing primer and variable 3’ ends depending on where the dideoxynucleotide has been inserted.
Traditional dideoxy sequencing methods employed radioisotope labelling (35S labelled oligonucleotides) using a dNTP mix that contains a proportion of radiolabel led nucleotides, which are incorporated within the growing DNA chains. Size fractionation of products of the four reactions is carried out in separate wells of a polyacrylamide gel. The gel is dried and subjected to autoradiography allowing the complementary strand to be read from top to bottom. Figure.7 shows an example of a sequence within the gene for neurofibromatosis type-1.
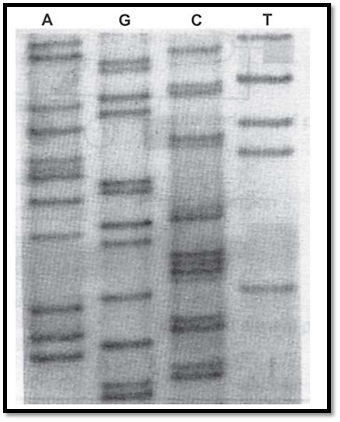
Fig. 7 : A portion of the sequencing gel showing the nucleotide sequence of a single stranded DNA template from the neurofibromatosis type-1 gene
Cycle Sequencing
Cycle sequencing is also called linear amplification sequencing. It is a PCR sequencing approach, which uses a thermostable DNA polymerase and a temperature cycling format of denaturation, annealing and DNA synthesis. However, cycle sequencing employs only one primer and includes a ddNTP chain terminator in the reaction. Therefore the product accumulates linearly instead of exponentially as seen in a conventional PCR reaction. Double stranded plasmids, cosmids, and PCR products can be sequenced using this method.
Automated DNA Sequencing Using Fluorescent Labelling Systems
These procedures use primers or dideoxynucleotides, which have attached chemical groups called flurophores, which are capable of fluorescing. Different flurophores are used for the four base specific reactions, and therefore all four reactions are loaded in a single lane. During electrophoresis a monitor detects and records a fluorescent signal as the DNA passes through a fixed point in the gel. As individual fragments migrate past this position, the laser causes the dyes to fluoresce. Maximum fluorescence occurs at different wavelengths for the four dyes and the information is recorded electronically. An example of automated DNA sequence using fluorescent primers is shown in Figure.8.

Fig. 8: Automated DNA sequencing using fluorescent primers showing output of sequence data from an automated DNA sequencer and dye and basic specific probes
References
Purandarey , H. (2009) . Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
تفوقت في الاختبار على الجميع.. فاكهة "خارقة" في عالم التغذية
|
|
|
|
|
|
|
أمين عام أوبك: النفط الخام والغاز الطبيعي "هبة من الله"
|
|
|
|
|
|
|
خدمات متعددة يقدمها قسم الشؤون الخدمية للزائرين
|
|
|
















