آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| CANCER GENETICS |


|
|
Read More
Date: 12-11-2015
Date: 11-11-2015
Date: 10-11-2015
|
CANCER GENETICS
INTRODUCTION
A neoplasm is an abnormal tissue, which grows when normal cellular control mechanisms fail. A neoplasm can involve any tissue of the body and may be benign or malignant. The etiology of most neoplasms is multifactorial. Both inherited and noninherited factors are involved in the pathogenesis. Non-inherited factors are genetic somatic mutations, which act as the main components in the development of a neoplastic process. Some malignant neoplasms have predisposing factors, which are inherited as Mendelian traits. In these cases neoplasms have an earlier age of onset. The specific abnormalities are characteristic of specific Mendelian patterns, suggesting that some predisposing factors responsible for neoplasm are heritable.
MENDELIAN TRAITS
a. Autosomal dominant traits include conditions such as neurofibromatosis type I, multiple endocrine neoplasia, inherited breast cancer, familial polyposis coli, and hereditary non-polyposis colon cancer (HNPCC).
b. Autosomal recessive traits are seen in abnormalities of DNA or chromosome repair. Affected individuals show an increased frequency of abnormal DNA repair or increased chromosomal breakage. Some examples of these are xeroderma pigmentosum, Fanconi’s anemia, and ataxia telangectasia.
c. Sex linked traits include the immunodeficiency syndromes. In this group affected individuals have congenital abnormalities of immunologic function and traits, which are inherited as autosomal or sex linked. Examples include X-linked agammaglobulinemia, and Wiskott-Aldrich syndrome.
Some examples of single gene disorders with cancer as a complication include skin carcinomas in xeroderma pigmentosum, lymphomas in ataxia telangiectasia, acute leukemia’s in Down syndrome, renal embryonic tumors in Wilm’s tumor, colonic adenocarcinomas in familial adenomatosis polyposis, breast carcinomas in hereditary breast cancer, cerebellar hemangioblastomas and renal carcinomas in Von Hippel Lindau disease, fibrosarcomas and optic gliomas in neurofibromatosis type 1, acoustic neuromas, schwannomas and meningiomas in neurofibromatosis type 2, retinal embryonic tumors in retinoblastoma, renal angiomyolipomas, cardiac rhabdomyomata and giant cell astrocytomas in tuberous sclerosis, pheochromocytoma and medullary thyroid carcinoma in multiple endocrine neoplasia type Ila and Ilb.
Chromosome abnormalities and neoplasia
Constitutional Chromosomal Abnormalities
Some constitutional chromosome abnormalities (i.e. inherited abnormalities that are present in every cell of the body) show an increased frequency of certain kind of malignancies in a proportion of patients. Examples of these include leukemia in patients with Down syndrome, retinoblastomas in patients with a, deletion of chromosome 13 (13q14.1) (Fig. 1) and Wilms tumor in patients with a deletion of chromosome 11p13.

Fig. 1: Karyotype of a patient with retinoblastoma. The arrow shows short chromosome 13 due to interstitial deletion of 13q 14
Acquired Chromosome Abnormalities
Each tumor results from one or more mutations of cellular DNA. These are seen in most malignant neoplasms. Patients do not have a constitutional chromosomal abnormality; instead, the tumor tissue acquires mutations. An example of this is chronic myelogenous leukaemia (CML). The chromosome abnormalities noted is a translocation between chromosomes 9 and 22, der(22)t(9;22),(Fig. 2) commonly known as Philadelphia chromosome.
Certain cells exhibit different karyotypic abnormalities in a single neoplasm, but the progenitor may be the same. As the neoplasm progresses, the karyotype tends to become more abnormal. Karyotype abnormalities usually precede clinical sign of relapse.
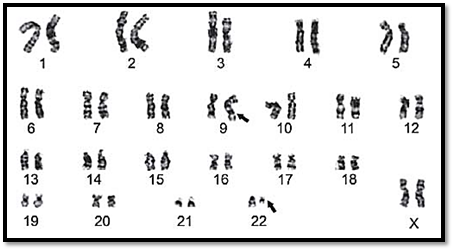
Fig. 2: Karyotype of a patient with chronic myeloid leukaemia. A part of chromosome 22 is translocated on to the long arm of chromosome number 9
Clonal Origin
The majority of malignant neoplasms are of clonal origin. That means all of the neoplastic cells originate from a single progenitor, which is abnormal. Malignancies occur as a multistep neoplastic process.
Telomeres and Cancer
After a fixed number of cell divisions, normal cells become arrested in a terminally non-dividing state known as cellular senescence. With each cell division, there is shortening of the telomeres at the ends of chromosomes. Once the chromosomes are shortened beyond a certain point, the loss of telomere function leads to end-to-end chromosome fusion and cell death. In germ cells, telomere shortening is prevented by the sustained function of the enzyme telomerase, thus explaining the ability of these cells to self-replicate extensively. Telomerase is absent from most somatic cells and hencethey suffer progressive loss of telomeres. In cancer cells, telomerase activity has been detected in a vast majority of human tumors.
The role of the environment and genes in cancer
Molecular studies have confirmed the genetic basis of both sporadic and heritable cancer.
The role of the environment in the development of cancer has also been clearly established. The role of factors such as chemical carcinogens, radiation energy, chronic irritation, hormones, parasites and oncogenic viruses in the development of cancer has also been established. Environmental predisposing factors leading to cancer may be occupational, like prolonged exposure to carcinogenic chemicals. Examples include bladder cancer in aniline dye workers, lung cancer in asbestos workers or skin cancer in tar workers. Genetic predisposition such as heredity, age, pigmentation, sex, and tumor immunity are also responsible factors. Cancers like those of the breast, ovary and colon are known to have a strong familial predisposition.
Predisposing genetic factors in cancer
Carcinogenesis is a multistep process, at both the phenotypic and genetic levels. Phenotypically, a malignant neoplasm shows excessive growth, local invasiveness and ability to metastasise. At the molecular level, accumulation of mutations in genes and chromosomes result in tumor progression.
In cancer genetics, genes acting as predisposing factors for carcinogenesis are grouped into four main classes. These groups are growth-promoting oncogenes, tumor suppressor genes, genes that regulate programmed cell death or apoptosis and DNA repair genes.
Oncogenes
Oncogenes are derived from proto-oncogenes, which are cellular genes that promote normal growth and differentiation. They were first recognised, as genes of tumor causing viruses that are responsible for the process of transformation. v-onc denotes viral oncogene and c-onc denotes cellular counterpart of a viral oncogene. Proto-oncogene conversion to oncogenes commonly occurs at the somatic level and causes sporadic cancers. A known germline mutation in the ret oncogene is responsible for multiple endocrine neoplasia type IIa, which is inherited as a dominant pattern.
Oncogene products can be grouped into five classes:
1- Protein tyrosine kinases such as abl (Abelson murine leukemia virus) and src (Rous/avian sarcoma virus)
2- Growth factors such as sis (simian sarcoma virus)
3- Growth factor receptors such as erbB (avian erythroblastosis virus)
4- Guanyl nucleotide binding proteins or G proteins such as H-ras (Harvey murine sarcoma virus) and K-ras (Kirsten murine sarcoma virus)
5- Transcription factors/DNA binding proteins such as myc (avian myelocytomatosis virus) and fos (FBJ osteosarcoma virus).
In tumor cells, oncogenes act in a dominant fashion. Mechanisms by which proto-oncogenes become oncogenes include:
1- Point mutations: A large number of human tumors carry point mutations in the ras group of oncogenes.
2- Chromosomal rearrangements including translocations: Alteration in chromosome number and structure is well documented in leukemia’s lymphomas, and solid tumors. Certain chromosomes are more involved and are specific to the type of malignancy. Chromosomal translocations result in rearrangements in the vicinity of the region of protooncogenes. These rearrangements lead to chimeric fusion and alteration in biochemical functions. Over 100 translocations are associated with carcinogenesis. An example of translocation is in Burkitt’s lymphoma, where there is a translocation of the c-myc gene from chromosome 8 to chromosome 14 next to the immunoglobulin heavy chain gene. Another example of an oncogene activated by translocation is in chronic myeloid leukemia, where there is formation of the Philadelphia chromosome. This is a reciprocal translocation between chromosomes 9 and 22 where a portion of the c-abl oncogene from chromosome 9 is translocated to bcr on chromosome 22.
3-Gene amplification: Activation of proto-oncogenes may result in amplification of their DNA sequences. In some cases the amplified genes produce cytogenetic changes that can be seen microscopically, called double minutes (dms) or homogenous staining regions (HSRs). Examples of gene amplification include the n-myc gene in neuroblastoma and the c-erbB2 gene in breast cancers.
Tumor suppressor genes
Normal cells contain genes with tumor suppressor activity, which when lost or inactivated can lead to malignancy.
Tumor suppressor genes include the following categories:
1- Molecules that regulate nuclear transcription and the cell cycle. These include the retinoblastoma gene (Rb), and the p53 gene.
2- Molecules that regulate signal transduction. These include the products of the gene for neurofibromatosis type-1 (NF1) and the gene for familial adenomatosis coli (APC)
3- Other tumor suppressor genes. These include genes for neurofibromatosis type 2 (NF2), the gene for von Hippel Lindau disease (VHL), and the gene for Wilms’ tumor (WT-1).
Some of the disorders caused by these genes are discussed in more detail below.
Retinoblastoma (Rb)
This is a rare highly malignant disorder of the retinal cells and leads to blindness and death if undetected. It affects approximately 1 in 20,000 children. Unilateral tumors are mostly sporadic while bilateral tumors are hereditary, in which mutation or loss of both normal copies of Rb genes are required to produce retinoblastoma. Bilateral tumors, which are hereditary, follow Knudsen’s two hit hypothesis. This hypothesis states, that in bilateral tumors, the first mutation is non functional and present in all the cells while a second gene at the same locus becomes inactivated somatically in the developing retina. This suggests that the retinoblastoma gene acts recessively as a tumor suppressor gene. The mode of inheritance for retinoblastoma is autosomal dominant with incomplete penetrance. 80-90% of children who inherit the autosomal gens develop retinoblastoma. 5% of children with retinoblastoma have some associated physical abnormalities. Cytogenetic analyses of affected children show an interstitial deletion of the long arm of one of the pair of chromosome 13 at 13q14.
Wilms Tumor
Wilms’ tumor commonly occurs unilaterally and its occurrence is sporadic. 20% of bilateral tumors show hereditary occurrence in at least 1% of the cases. The clinical features include aniridia, genitourinary abnormalities and mental retardation in a few cases. Some cases show identification of an interstitial deletion of chromosome 11 (11p13) and the gene WT1 acts as a tumor suppressor gene.
von Hippel Lindau Disease (VHL)
It is inherited as a multisystem disorder characterized by abnormal growth of blood vessels. This results in the development of hemangioblastomas throughout the brain, especially in the cerebellum, retina and spinal cord. Renal, hepatic, adrenal and pancreatic cysts are known to occur. The VHL gene on chromosome 3 is a tumor suppressor gene and is inherited in an autosomal dominant fashion. If the gene is lost or mutated then its inhibitory effect on cell growth is lost or diminished which in combination with defects in other regulatory proteins can lead to cancerous growths.
Colorectal Cancer
Colorectal tumors progress through a series of stages ranging from benign adenomatous polyps to malignant carcinomas. This progression is the result of a series of genetic changes that involve activation of oncogenes and inactivation of tumor suppressor genes. Colorectal cancer typifies the multistep nature of the biology and pathogenesis of cancers in general. In general, colorectal carcinoma is thought to originate mainly from adenomas. A combination of at least six genetic events are involved in the pathogenesis of colorectal cancer. The initial event is a germline or somatic mutation in the APC gene, a tumor suppressor gene located on 5q21. The next stage involves loss of heterozygosity (LOH) of the second APC gene. Activation of two recessive oncogenes (Ras genes, KRAS1 on chromosome 6p12-11 and KRAS2 on chromosome 12p12) has been associated with this transformation. Loss of the DCC (deleted in colorectal carcinoma) tumor suppressor genes, located at 18q21.3, and mutation in the DNA sequences at 5q21-22 (MCC, mutated in colorectal carcinoma) are also seen. The final step into a cancerous state is the result of mutations in the tumor suppressor gene, p53 located on chromosome 17p12- 13. The transition to metastatic carcinoma involves mutations in the two RAS genes, KRAS1 and KRAS2. The exact order of genetic changes described above varies, although some mutations occur more frequently as an early event, while other mutations typically happen later in the process. It is the accumulation of genetic mutations, which is important in colorectal cancer development.
Of all colon cancers that are diagnosed in the United States, most are sporadic, with no family history of colon cancer. However, up to 10% of colon cancers are thought to be due to an inherited gene mutation that may be passed directly from generation to generation. Familial adenomatosis polyposis (FAP) and hereditary non-polyposis coli (HNPCC) are types of inherited colon cancer.
FAP is inherited as an autosomal dominant disorder and carries a high risk of malignancy. Patients with FAP develop hundreds of benign colorectal tumors, some of which will progress to carcinomas. FAP is associated with germline mutations of the APC gene on chromosome 5q. In addition, the APC gene mutations also occur somatically in over 60% sporadic colorectal tumors. Mutations in the APC gene can be identified in tumors as small as 0.5 cm in diameter.
HNPCC is also inherited in an autosomal dominant manner. The genes implicated in the genetic susceptibility to HNPCC are called mismatch repair genes or MMR genes. These genes act as spellcheckers to ensure that the sequence of DNA is correct as genes are duplicated during the cell cycle. The MMR genes are responsible for repairing mistakes in the DNA sequence so that each new cell receives a correct set of genes. Within HNPCC families, mutations in four different MMR genes have been identified. Mutation in the gene, hMSH2, located on chromosome 2, accounts for 31% of HNPCC families. Mutation in the gene, hMLH1 , located on chromosome 3, account for 33% of HNPCC families. Mutations in the hPMS1 (chromosome 2) and hPMS2 (chromosome 7) genes appear to account for 2% and 4% of HNPCC families, respectively.
Breast Cancer
Breast cancer is the most frequently diagnosed cancer in Western women. Numerous risk factors for the development of breast cancer have been identified. 20% of women with breast cancer have a family history of the disease, and 5% of these are attributable to mutations in two genes. Mutations in the BRCA1 or BRCA2 genes increase susceptibility to develop breast and/ or ovarian cancer. The BRCA1 gene is on chromosome 17q21 and families with germ-line mutations in BRCA1 have an autosomal dominant inheritance in the pattern of breast cancer as well an increased incidence of ovarian cancer. Mutations in BRCA1 account for 20-30% of the inherited breast cancers. The BRCA2 gene is located on chromosome 13q12-13. Families with germ-line mutations in BRCA2 account for 10-20% of inherited breast cancers. The pattern of breast cancer inheritance for BRCA2 is autosomal dominant, an increased incidence of ovarian cancer that is less striking than that with BRCA1 and an increased incidence of male breast cancer.
Many somatic mutations are believed to be responsible for the development of breast cancer. Oncogenes erbB1, erbB2, myc and int2 are responsible for the malignancy. Loss of heterozygosity for a number of chromosomes for example 1q, 3p, 11p, and 13q is also seen.
Neurofibromatosis Types 1 and 2
Neurofibromatosis type 1 (NF1) also known as von Recklinghausen’s disease and has an incidence of 1:3000. It is commonly inherited in an autosomal dominant manner. Patients present with cafe au lait patches, cutaneous neurofibromas, Lisch nodules of the iris, axillary freckling and optic gliomas. 5% of the affected people show malignancies including neurofibrosarcomas and embryonal tumors. The gene for NF1 is cloned and islocalized to chromosome 17q11.2.
Neurofibromatosis type 2 (NF2) is also a dominantly inherited condition. Patients have bilateral acoustic neuromas (vestibular schwannomas). 40% of the patients have spinal and intracranial tumors. The prognosis of NF2 is poor as surgical removal of the VIII th nerve tumor is difficult. The NF2 gene is located on chromosome 22. Presymptomatic screening and prenatal diagnosis in affected families is possible.
Genes that regulate apoptosis
Genes that prevent or induce programmed cell death or apoptosis also play a role in cancer. Examples of genes that regulate apoptosis include the bcl-2 gene, which inhibits apoptosis and the bax, and bad genes that favour programmed cell death. Two other cancer-associated genes that are also connected with apoptosis are the p53 gene and the c-myc proto oncogene.
Genes that regulate DNA repair
DNA repair genes themselves are not oncogenic but allow mutations in other genes during the process of normal cell division. Individuals born with inherited mutations of DNA repair proteins are at greatly increased risk of developing cancer.
Examples of such diseases are Hereditary non-polyposis coli (HNPCC), in which of the various DNA mismatch repair genes, mutations in hMSH2 on chromosome 2 account for tumor development in 50% of families. Microsatellite instability is the hallmark of defective mismatch repair. Other examples include xeroderma pigmentosum, Bloom’s syndrome, ataxia telangiectasia and Fanconi’s anemia (These are discussed in detail in the chapter on chromosomal syndromes).
Ovarian Cancer
Ovarian cancer is a potentially lethal neoplasm of the female reproductive system. It is observed that 80% of malignant tumors of the ovary originate from the surface epithelium. Several molecular events such as loss of heterozygosity (LOH) at different sites on chromosomes 6, 11,13 and 17, mutations in tumor suppressor genes (such as p53, BRCA1, BRCA2), and mutation of proto-oncogenes (RAS, FOS and MYC) are responsible for development of ovarian tumors. Majority of ovarian cancers are of sporadic in origin. Diagnosing ovarian cancer at on early stage is difficult due to the large space available for the ovaries to grow, as a result patients are asymptomatic in the early stages and by the time it is detected it reaches incurable stage. Transvaginal sonography and colour Doppler is recommended annually in high-risk cases.
Screening for familial cancer
As of today the standard management for cancer is prevention and early detection. Prevention aspects include diet, change in life style, drugs, prophylactic surgery or screening. Screening is carried out at early stage, before any phenotypic expression occurs. For example, in individuals at risk for familial adenomatous polyposis screening is by endoscopic examination. Retinal examination for congenital hypertrophy of the retinal pigment epithelium in family with a history of retinoblastoma is worthwhile. Presymptomatic screening should be considered in high-risk families.
The test should be sensitive and specific enough to pick a malignant or pre-malignant condition prior to producing symptoms. Screened persons should benefit, due to early detection resulting in improved prognosis. Benefits from the test should be more than the potential dangers. It should be non-invasive whenever possible. Appropriate pre-screening counselling and facilities for follow up of the cases should be available. In colorectal cancer endoscopy provides a sensitive test that can be offered to at risk patient. In breast cancer regular mammography after the age of 40 years at intervals of five years in women who are at risk for breast cancer is ideal and is a national program in the United Kingdom. Breast self-examination or clinical examination should be encouraged as a preventive early screening.
Recent developments in biotechnology in cancer therapeutics
Some of the Techniques Used in the Treatment of Cancer Biotechnology are Discussed
Antisense Technology
Antisense methods involve the disruption of gene expression using short, sequence specific DNA molecules called oligonucleotides. These synthesized oligonucleotides bind complementary DNA molecules in the double helix of the genome, and form triplexes that prevent ribosomal protein synthesis by prohibiting the translation process of mRNA.
Recombinant Retroviral Vectors
Retroviral mediated gene transfer has also been used to incorporate antisense vectors into cancerous cells. The vector will produce complementary RNA sequences that combine with the target RNA sequence, resulting in a double strand of RNA, which will be degraded by intracellular enzymes, instead of being expressed through ribozymes.
Gene Therapy
In this technique, recombinant vectors are used so that they will infect specific cells of interest. There are many viruses with different predilection sites in body, which affect only specific cells. These vectors will transfect specific cells and express genes, which will induce an immune response to malignant cells. There are several mechanisms involved by which cancer cells are able to escape from immune surveillance. Recombinant retrovirus mediated gene transfer of vectors expressing interferon Y can boost the expression of MHC I and II proteins which induce strong cytotoxic T cell immune response and inhibit the growth of cancer cells.
Drug Resistance Genes
In chemotherapy, the main hurdle is side effects such as hemotoxicity. Recombinant-mediated gene transfer of drug resistance genes such as multi-drug resistance gene (MDR1), mutant dihydrofolate reductase genes, and methyl-transferase genes to the bone marrow stem cells have the potential to produce drug tolerance to toxic drugs. This may permit an increased dose of the drugs.
Gene Chip Technology
The gene chip is also referred to as DNA chip array technology. The gene chip is a glass wafer, which is approximately 2cm2 in size. On top of this glass, several arrays of known nucleotide sequences of oligonucleotide probes are arranged. This glass chip is encased into black plastic cartridges in which the reaction takes place. The main principle of this technology is the ability of nucleic acids to hybridise with complementary DNA sequences with high stringency. For example, DNA with sequence TTGGCAT will hybridise with AACCGTA nucleotide sequence.
If there is single nucleotide, which is not complementary, then it will reduce the binding affinity with a low signal of fluorescence. This technology can be exploited to detect well- characterized mutations in cancer. A person with one type of cancer showing a similar phenotype may show different mutations in candidate genes. It may be a point mutation, deletion, insertion or base substitution. (Based on types of mutations in candidate genes, it may be possible to design effective drugs) Using the gene chip technology, it is possible to test more than 600 mutations of the CFTR gene responsible for cystic fibrosis, on a single chip. Routine genetic techniques to detect mutations are difficult, laborious, time consuming and at times may be inconclusive. This DNA chip technology will also be used for clinical surveillance of presence of cancer genes or cancer associated mutations in affected tissues at an early age, in patients who are predisposed for developing specific cancers before it gets expressed in later life. Accordingly, preventive or prophylactic therapeutic strategies can be recommended for the high-risk patient.
References
Purandarey, H. (2009). Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
علامات بسيطة في جسدك قد تنذر بمرض "قاتل"
|
|
|
|
|
|
|
أول صور ثلاثية الأبعاد للغدة الزعترية البشرية
|
|
|
|
|
|
|
مكتبة أمّ البنين النسويّة تصدر العدد 212 من مجلّة رياض الزهراء (عليها السلام)
|
|
|