آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| PCR and RT-PCR |


|
|
Read More
Date: 29-12-2015
Date: 1-5-2016
Date: 16-12-2015
|
PCR and RT-PCR
KEY CONCEPTS
-Polymerase chain reaction permits the exponential amplification of a desired sequence by using primers that anneal to the sequence of interest.
-RT-PCR uses reverse transcriptase to convert RNA to DNA for use in a polymerase chain reaction.
-Real-time, or quantitative, polymerase chain reaction detects the products of PCR amplification during their synthesis, and is more sensitive and quantitative than conventional PCR.
- PCR depends on the use of thermostable DNA polymerases that can withstand multiple cycles of template denaturation.
Few advances in the life sciences have had the broad-reaching and even paradigm-shifting impact of the polymerase chain reaction (PCR). Although evidence exists that the underlying core principles of the method were understood and in fact used in practice by a few isolated people prior to 1983, credit for independent conceptualization of the mature technology and foresight of its applications must go to Kary Mullis, who was awarded the 1993 Nobel Prize in Chemistry for his insight.
The underlying concepts are simple and based on the knowledge that DNA polymerases require a template strand with an annealed primer containing a 3′ hydroxyl to commence strand extension. The steps of PCR are illustrated in FIGURE 1. While in the context of normal cellular DNA replication this primer is in the form of a short RNA molecule provided by DNA primase, it can equally well be provided in the form of a short, single-stranded synthetic DNA oligonucleotide having a defined sequence complementary to the 3′ end of any known sequence of interest. Heating of the double-stranded target sequence of interest (known as the “template molecule,” or just “template” for short) to near 100°C in an appropriate buffer causes thermal denaturation as the template strands melt apart from each other (Figure .1a and b). Rapid cooling to the annealing temperature (or Tm ) of the primer/template pair and a vast molar excess of the short, kinetically active synthetic primer ensures that a primer molecule finds and appropriately anneals to its complementary target sequence more rapidly than the original opposing strand can do so (Figure .1c). If presented to a polymerase, this annealed primer presents a defined location from which to commence primer extension (Figure .1d). In general, this extension will occur until either the polymerase is forced off the template or it reaches the 5′ end of the template molecule and effectively runs out of template to copy.
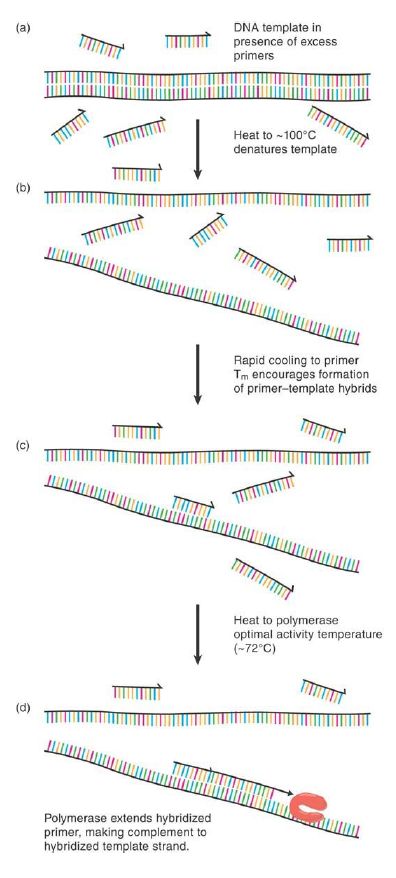
FIGURE .1 Denaturation (a) and rapid cooling (b) of a DNA template molecule in the presence of excess primer allow the primer to hybridize to any complementary sequence region of the template (c). This provides a substrate for polymerase action and primer extension (d), creating a complementary copy of one template strand downstream from the primer.
The ingenuity of PCR arises from simultaneously incorporating a nearby second primer of opposing polarity (i.e., complementary to the opposite strand to which the first primer anneals) and then subjecting the mixture of template, two primers (at high concentrations), thermostable DNA polymerase, and dNTP containing polymerase buffer to repeated cycles of thermal denaturation, annealing, and primer extension. Consider just the first cycle of the process: Denaturation and annealing occur as described earlier, but with both primers, creating the situation depicted in FIGURE .2 If polymerase extension is allowed to proceed for a short period of time (on the order of 1 minute per 1,000 base pairs), each of the primers will be extended out and past the location of the other, thus creating a new complementary annealing site for the opposing primer. Raising the temperature back to denaturation stops the primer elongation process and displaces the polymerases and newly created strands. As the system is cooled again to the annealing temperature, each of the newly formed short, single DNA strands serves as an annealing site for its opposite polarity primer. In this second thermal cycle, extension of the primers proceeds only as far as the template exists—that is, the 5′ end of the opposing primer sequence. The process has now made both strands of the short, defined, precisely primer-to-primer DNA sequence. Repeating the thermal steps of denaturation, annealing, and primer extension leads to an exponential increase (2N , where N is the number of thermal cycles) in the number of this defined product, allowing for phenomenal levels of “sequence amplification.” Close consideration of the process reveals that even though this also creates uncertain length products from the extension of each primer off the original template molecule with each cycle, these products accrue in a linear fashion and are quickly vastly outnumbered by the primer-to-primer defined product, known as the amplicon. In fact, within 40 thermal cycles of an idealized PCR reaction, a single template DNA molecule generates approximately 1012 amplicons—more than enough to go from an invisible target to a clearly visible fluorescent dye–stained product.
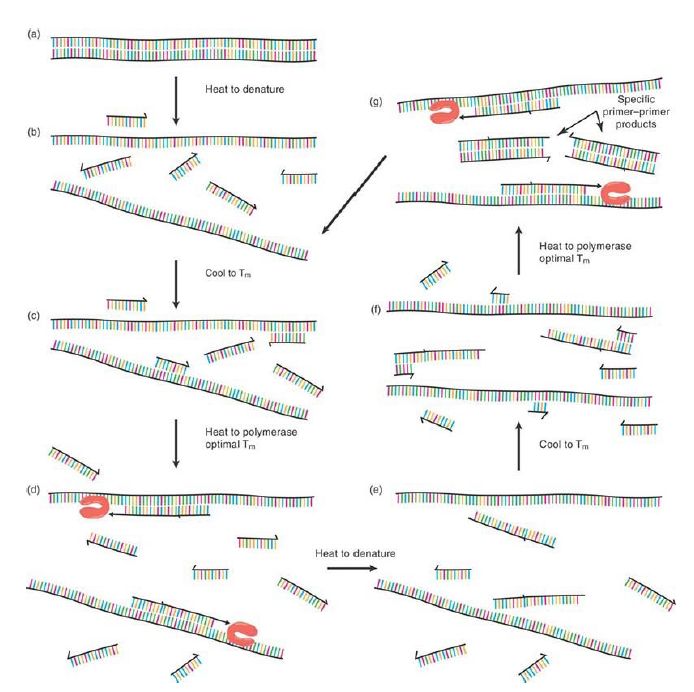
FIGURE 2. Thermally driven cycles of primer extension where primers of opposite polarity have nearby priming sites on each of the two template strands lead to the exponential production of the short, primer-to-primer–defined sequence (the “amplicon”).
Perhaps not surprisingly, there are many technical complexities underlying this deceptively simple description. Primer design must take into account issues such as DNA secondary structures, uniqueness of sequence, and similarity of Tm between primers. Use of a thermostable polymerase (that is, one that is not inactivated by the high temperatures used in the denaturation steps) is an essential concept identified by Mullis and coworkers. Within this constraint, however, different enzyme sources with differing properties (e.g., exonuclease activities for increased accuracy) can be exploited to meet individual application needs. Buffer
composition (including agents such as DMSO to help reduce secondary structural barriers to effective amplification, and inclusion of divalent cations such as Mg2+ at sufficient concentration not to be depleted by chelation to nucleotides) often needs some optimization for effective reactions. In general, the PCR process works best when the primers are within short distances of each other (100 to 500 base pairs), but well optimized reactions have been successful at distances into the tens of kilobases. “Hot start” techniques—frequently through covalent modification of the polymerase—can be employed to ensure that no inappropriate primer annealing and extension can occur prior to the first denaturation step, thereby avoiding the production of incorrect products. Generally, somewhere around 40 thermal cycles marks an effective limit for a PCR reaction with good kinetics in the presence of appropriate template, as depletion of dNTPs into amplicons effectively occurs around this point and a “plateau phase” occurs wherein no more product is made. Conversely, if the appropriate template was not present in the reaction, proceeding beyond 40 cycles primarily increases the likelihood of production of rare, incorrect products.
Pairing PCR with a preliminary reverse transcription step (either random-primed or using one of the PCR primers to direct activity of the RNA-dependent DNA polymerase [reverse transcriptase]) allows for RNA templates to be converted to cDNA and then subject to regular PCR, in a variation known as reverse transcription PCR (RT-PCR). In general, the subsequent discussion uses the term PCR to refer to both PCR and RT-PCR.
Detection of PCR products can be done in a number of ways. Postreaction “endpoint techniques” include gel electrophoresis and DNA-specific dye staining. Long a staple of molecular biological techniques , this is a simple but effective technique to rapidly visualize both that an amplicon was produced and that it is of an expected size. If the particular application requires exact, to-thenucleotide product sizing, capillary electrophoresis can be used instead. Hybridization of PCR products to microarrays or suspension bead arrays can be used to detect specific amplicons when more than one product sequence might come out of an
assay. These in turn use a variety of methods for amplicon labeling, including chemiluminescence, fluorescence, and electrochemical techniques. Alternatively, real-time PCR methodologies employ some way of directly detecting the ongoing production of amplicons in the reaction vessel, most commonly through monitoring a direct or indirect fluorescence change linked to amplicon production by optical methods. These methods allow the reaction vessel to stay sealed throughout the process. In contrast to endpoint methods for which final amplicon concentration bears little relationship to starting template concentration, real-time methods show good correlations between the thermocycle number at which clear signals are measurable—usually referred to as the threshold cycle(C )—and the starting template concentration. Thus, realtime methods are effective template quantification approaches. As a result, these methods are often referred to as quantitative PCR (qPCR) methods.
Conceptually, the simplest method for real-time PCR detection is based on the use of dyes that selectively bind and become fluorescent in the presence of double-stranded DNA, such as SYBR green. Production of a PCR product during thermocycling leads to an exponential increase in the amount of double-stranded product present at the annealing and extension thermal steps of each cycle. The real-time instrument monitors fluorescence in each reaction tube during these thermal steps of each cycle and calculates the change in fluorescence per cycle to generate a sigmoidal amplification curve. A cutoff threshold value placed approximately midrange in the exponential phase of this curve is used for calculating the CT of each sample and can be used for quantitation if appropriate controls are present.
A potential issue with this approach is that the reporter dyes are not sequence specific, so any spurious products produced by the reaction can lead to false-positive signals. In practice, this is
usually controlled for by performance of a melt point analysis at the end of regular thermocycling. The reaction is cooled to the annealing temperature, and then the temperature is slowly raised while fluorescence is constantly monitored. Specific amplicons will have a characteristic melt point at which fluorescence is lost, whereas nonspecific amplicons will demonstrate a broad range of melt points, giving a gradual loss in sample fluorescence.
A number of alternate approaches use probe-based fluorescence reporters, which avoid this potential nonspecific signal. Probebased approaches work through the application of a process called fluorescence resonant energy transfer (FRET). In simple terms, FRET occurs when two fluorophores are in close proximity and the emission wavelength of one (the reporter) matches the excitation wavelength of the other (the quencher). Photons emitted at the reporter dye emission wavelength are effectively captured by the nearby quencher dye and reemitted at the quencher emission wavelength. In the simplest form of this approach, two short oligonucleotide probes with homology to adjoining sequences within the expected amplicon are included in the assay reaction; one probe carries the reporter dye, and the other the quencher. If specific PCR product is formed in the reaction, at each annealing step these two probes can anneal to the single-stranded product and thereby place the reporter and quencher molecules close to each other. Illumination of the reaction with the excitation wavelength of the reporter dye will lead to FRET and fluorescence at the quencher dye’s characteristic emission frequency. By contrast, if the homologous template for the probe molecules is not present (i.e., the expected PCR product), the two dyes will not be colocalized and excitation of the reporter dye will lead to fluorescence at its emission frequency. This is illustrated in FIGURE 3. As with the DNA-binding dye approach, the real-time instrument monitors the quencher emission wavelength during each cycle and generates a similar sigmoidal amplification curve. Multiple alternate ways of exploiting FRET for this process exist, including 5′ fluorogenic nuclease assays, molecular beacons, and molecular scorpions. Although the details of these differ, the underlying concept is similar and all generate data in a similar fashion.
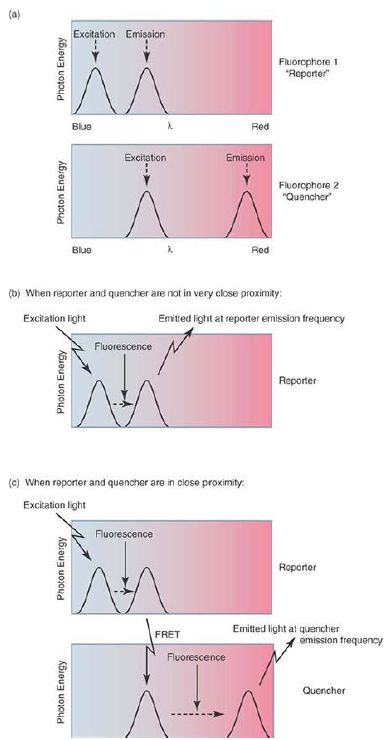
FIGURE 3. Fluorescence resonant energy transfer (FRET) occurs only when the reporter and quencher fluorophores are very close to each other, leading to the detection of light at the quencher emission frequency when the reporter is stimulated by light of its excitation frequency. If the reporter and quencher are not colocalized, stimulation of the reporter instead leads to detection of light at the reporter emission frequency. By placing the reporter and quencher fluorophores on single-stranded nucleic acid probes complementary to the expected amplicon, different variations on this method can be designed such that the occurrence of FRET can be used to monitor the production of sequence-specific amplicons.
The applications of the PCR process are incredibly diverse. The simple appearance or nonappearance of an amplicon in a properly controlled reaction can be taken as evidence for the presence or absence, respectively, of the assay target template. This leads to medical applications such as the detection of infectious disease agents at sensitivities, specificities, and speeds much greater than alternate methods. Whereas the two primer sites must be of known sequence, the internal section can be any sequence of a general length, which leads directly to applications for which a PCR product for a region known to vary between species (or even between individuals) can be produced and subject to sequence analysis to identify the species (or individual identity, in the latter case) of the sample template. Coupled with single-molecule sensitivity, this has provided criminal forensics with tools powerful enough to identify individuals from residual DNA on crime scene evidence as small as cigarette butts, smudged fingerprints, or a single hair. Evolutionary biologists have made use of PCR to amplify DNA from wellpreserved samples, such as insects encased in amber millions of years old, with subsequent sequencing and phylogenetic analysis, yielding fascinating results on the continuity and evolution of life on Earth. Quantitative real-time approaches have applications in medicine (e.g., monitoring viral loads in transplant patients), research (e.g., examining transcriptional activation of a specific target gene in a single cell), or environmental monitoring (e.g., water purification quality control).
In general, PCR reactions are run with carefully optimized Tm values that maximize sensitivity and amplification kinetics while ensuring that primers will only anneal to their exact hybridization matches. Lowering the Tm of a PCR reaction—in effect, relaxing the reaction stringency and allowing primers to anneal to not quite perfect hybridization partners—has useful applications, as well, such as in searching a sample for an unknown sequence suspected
to be similar to a known one. This technique has been successfully employed for the discovery of new virus species, when primers matching a similar virus species are employed. Similarly, during a PCR-directed cloning of a gene or region of interest, planned mismatches in the primer sequence and slightly lowered Tm s can be used to introduce wanted mutations in a process called sitedirected mutagenesis. It’s possible to perform differential detection of single nucleotide polymorphisms (SNPs) , which can be directly indicative of particular genotypes or serve as surrogate linked markers for nearby genetic targets of interest, through the design of PCR primers with a 3′ terminal nucleotide specific to the expected polymorphism. At the optimal T , this final crucial nucleotide can only hybridize and provide a 3′ hydroxyl to the waiting polymerase if the matching single nucleotide polymorphism occurs. This process is known by several names, including amplification refractory mutation selection (ARMS) or allele-specific PCR extension (ASPE).
The PCR process described thus far has been restricted to amplification of a single target per reaction, or simplex PCR. Although this is the most common application, it is possible to combine multiple, independent PCR reactions into a single reaction, allowing for an experiment to query a single, minute specimen for the presence, absence, or possibly the amount of multiple unrelated sequences. This multiplex PCR is particularly useful in forensics applications and medical diagnostic situations, but entails rapidly increasing levels of complexity in ensuring that multiple primer sets do not have unwanted interactions that lead to
undesired false products. At best, multiplexing tends to result in loss of some sensitivity for each individual PCR due to effective competition between them for limited polymerase and nucleotides. A final point of interest to many students with regard to PCR is its consideration from a philosophical perspective. In practice, performance of this now incredibly pervasive method requires the use of a thermostable polymerase, as previously indicated. These polymerases (of which there are a number of varieties) primarily derive from bacterial DNA polymerases originally identified in extremophiles living in boiling hot springs and deep-sea volcanic thermal vents. Few people would have been likely to suspect that studying deep-sea thermal vent microbes would be of such direct importance in so many other aspects of science, including those that impact on their daily lives. These unexpected links between topics serve to highlight the importance of basic research on all manner of subjects; critical discoveries can come from the least expected avenues of exploration.

|
|
|
|
علامات بسيطة في جسدك قد تنذر بمرض "قاتل"
|
|
|
|
|
|
|
أول صور ثلاثية الأبعاد للغدة الزعترية البشرية
|
|
|
|
|
|
|
مكتبة أمّ البنين النسويّة تصدر العدد 212 من مجلّة رياض الزهراء (عليها السلام)
|
|
|