Carbonyl Compounds : H Abstraction, Bond Cleavage & Cycloaddition
The n → π* excited states of carbonyl compounds display a rich chemistry in their own right.
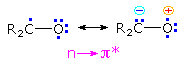
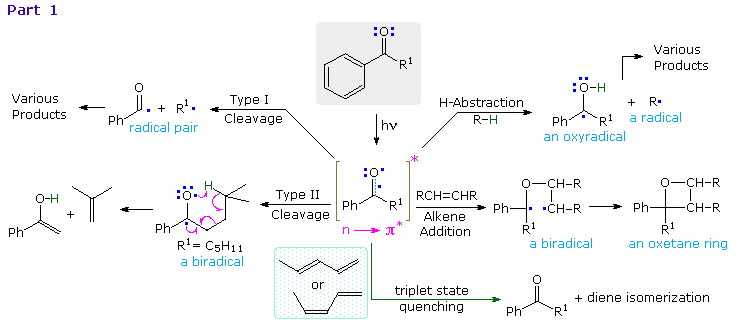
Since the oxygen has an unpaired electron, it behaves in much the same way as an alkoxy radical, as noted in the chapter on free radical chemistry. Hydrogen abstraction and addition to double bonds are typical reactions. Cleavage of neighboring carbon-carbon bonds may also occur, the two most common of these being designated Type I and Type II. General equations for these primary reactions, starting from aryl ketone excited states, are described in the following diagram. The n → π* excited state shown in brackets at the center of the diagram may also be described as the resonance hybrid drawn on the right. Note that the ground state polarity of the carbonyl group has been reversed in this excited state.
The triplet excited state of aryl ketones has a lifetime of about 100 ns, and most of the subsequent reactions are very fast (rates range from 104 to 109 M-1 sec-1). Consequently, the effect of quenchers such as 1,3-pentadiene on product distribution provides valuable information about the mechanisms. These quenchers have T1 energies much lower and S1 energies higher than corresponding ketone states. The rate of quenching is nearly diffusion controlled, and is therefore proportional to the concentration of the quencher. Since the ground state triplet of oxygen reacts rapidly with triplet excited states, air must be excluded when studying these reactions.
Examples of hydrogen abstraction and alkene addition reactions will be displayed above.
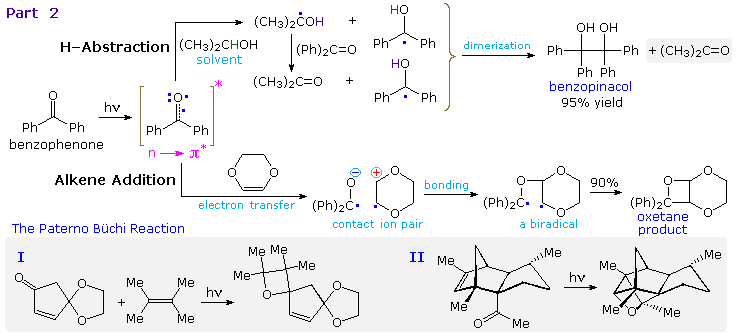
Reduction of benzophenone to benzpinacol is common in most hydrocarbon solvents, even in pure benzene. The diphenylmethanol radical formed by hydrogen abstraction is relatively stable and, once formed, dimerizes to benzopinacol in nearly quantitative yield. In contrast, the more reactive isopropanol radical is converted to acetone by transferring its hydroxyl hydrogen to benzophenone, thus forming another diphenylmethanol radical.
The photoaddition of carbonyl compounds to olefins had been observed by the Italian chemist E. Paterno in 1909, but the structure of the oxetane products remained unknown until elucidated by Büchi and coworkers (M.I.T.) in 1954. This reaction is now known as the Paterno-Büchi reaction. The mechanism shown here was established for the electron-rich alkene dioxene, a compound that favors electron transfer to benzophenone. Other alkenes are also believed to react by way of a 1,4-biradical intermediate, and significant regioselectivity is found with many unsymmetrical alkenes. Two additional examples of this reaction are drawn in the gray-shaded box at the bottom. The second illustrates the intramolecular variant. A related example of carbonyl addition to alkenes is the use of benzophenone as a light activated initiator of polymerization for monomers such as styrene and methyl methacrylate.
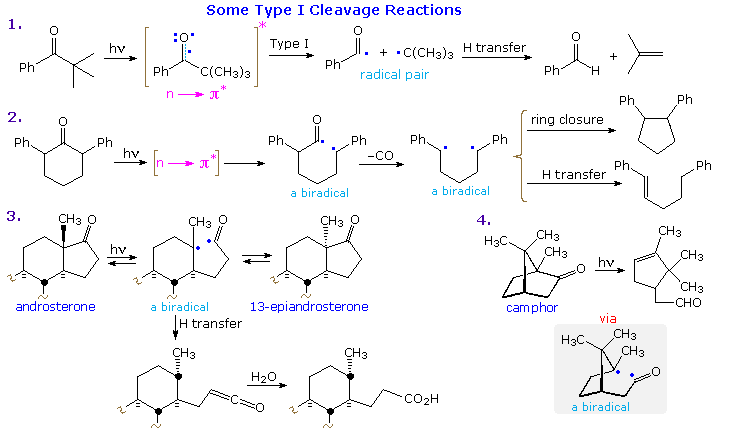
Examples of type I fragmentations are displayed below. These reactions are fast, and often proceed with a high quantum yield. The first example, shown at the top, illustrates a typical homolytic cleavage of a carbonyl substituent, followed by a fast hydrogen atom transfer between the caged radical pair. Three intramolecular type I reactions, all proceeding by way of biradical intermediates, demonstrate the variety of products that may be formed by hydrogen transfer disproportionation and/or ring closure. The cyclic transition states for such intramolecular hydrogen transfer usually include five or six, and occasionally seven atoms.
By clicking on the diagram, examples of type II reactions will be displayed. These reactions all begin by an intramolecular hydrogen atom abstraction, similar to the intermolecular abstraction in the reductive dimerization of benzophenone.The general example at the top of the diagram shows how a key 1,4-biradical intermediate, formed by intramolecular hydrogen abstraction, may disproportionate to several different products, including type II cleavage to the enol tautomer of a ketone (green shaded box) and ring closure to a cyclobutanol (the Yang Reaction). In hydrocarbon solvents intramolecular hydrogen transfer back to carbon is common, reducing the quantum yield of these products by over 50%. This reverse hydrogen transfer is inhibited by hydrogen bonding solvents, which stabilize the newly formed OH group. As expected, the rate at which the biradical is formed is sensitive to substitution at the γ-carbon. If R=H (a methyl group) the rate is 100 times slower than if R=CH3 (abstraction of a 3º- hydrogen). This reactivity order reflects the bond dissociation energies of the C–H bonds in the same fashion as noted for free radical halogenation of alkanes. Also, replacement of all γ-hydrogens by deuterium reduces the rate of biradical formation substantially.
The ratio of type II cleavage and Yang cyclization products from a given substrate depends on several factors. The steroid ketone in example 2 gives both reactions, with cyclization predominating. Example 3 is instructive, first because the rate of biradical formation is much higher than in many other cases, as a result of conformational bias favoring the reactive orientation (as drawn); and second, because cyclobutanol formation induces severe strain. Only type II fragmentation is observed. Finally, example 4 proceeds by a δ-hydrogen abstraction (7-membered cycle), as the only possible transformation of its kind. The resulting 1,5-biradical then closes to the product shown. Remarkably, phosphorescence studies indicate that this reaction occurs from a low-lying π→ π* triplet state, rather than the n→ π* triplet usually associated with this class of reaction.
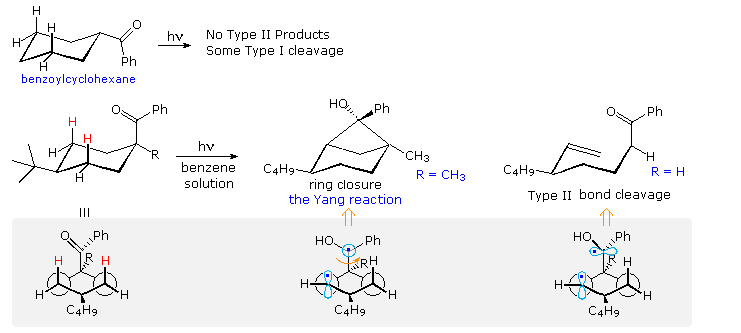
Type II reactions depend, of course, on close approach of the carbonyl oxygen to a γ-hydrogen. The conformational mobility of the examples drawn above permits this to occur within the lifetime of the excited triplet. However, it is not difficult to find molecules for which this is not possible. Benzoylcyclohexane, shown at the top of the following diagram, is such a compound. Because of its equatorial location on the six-membered ring, the carbonyl oxygen cannot reach any of the γ-hydrogens. If the benzoyl group is forced into an axial orientation by a large cis-tert-butyl group on c-4, then the oxygen can easily reach the axial cis γ-hydrogens (colored red. The resulting type II photochemistry depends on the other C-1 substituent. If R = H, type II cleavage is the only product; however this changes to exclusive stereoselective cyclization if R = CH3. The conformational drawings in the gray-shaded box suggest a reason for this remarkable change. Following the initial hydrogen abstraction, the 1,4-biradical is well aligned for ring closure, but the benzyl radical must be rotated 90º before it is properly aligned for bond cleavage (far right structure). A C-1 methyl substituent hinders this rotation, leading to formation of the strained cyclobutanol product.