























علم الكيمياء

تاريخ الكيمياء والعلماء المشاهير
التحاضير والتجارب الكيميائية
المخاطر والوقاية في الكيمياء
اخرى
مقالات متنوعة في علم الكيمياء
كيمياء عامة
الكيمياء التحليلية
مواضيع عامة في الكيمياء التحليلية
التحليل النوعي والكمي
التحليل الآلي (الطيفي)
طرق الفصل والتنقية
الكيمياء الحياتية
مواضيع عامة في الكيمياء الحياتية
الكاربوهيدرات
الاحماض الامينية والبروتينات
الانزيمات
الدهون
الاحماض النووية
الفيتامينات والمرافقات الانزيمية
الهرمونات
الكيمياء العضوية
مواضيع عامة في الكيمياء العضوية
الهايدروكاربونات
المركبات الوسطية وميكانيكيات التفاعلات العضوية
التشخيص العضوي
تجارب وتفاعلات في الكيمياء العضوية
الكيمياء الفيزيائية
مواضيع عامة في الكيمياء الفيزيائية
الكيمياء الحرارية
حركية التفاعلات الكيميائية
الكيمياء الكهربائية
الكيمياء اللاعضوية
مواضيع عامة في الكيمياء اللاعضوية
الجدول الدوري وخواص العناصر
نظريات التآصر الكيميائي
كيمياء العناصر الانتقالية ومركباتها المعقدة
مواضيع اخرى في الكيمياء
كيمياء النانو
الكيمياء السريرية
الكيمياء الطبية والدوائية
كيمياء الاغذية والنواتج الطبيعية
الكيمياء الجنائية
الكيمياء الصناعية
البترو كيمياويات
الكيمياء الخضراء
كيمياء البيئة
كيمياء البوليمرات
مواضيع عامة في الكيمياء الصناعية
الكيمياء التناسقية
الكيمياء الاشعاعية والنووية

Regioselective Enolate Formation and Diastereoselectivity
 المؤلف:
William Reusch
المؤلف:
William Reusch
 المصدر:
Virtual Textbook of Organic Chemistry
المصدر:
Virtual Textbook of Organic Chemistry
 الجزء والصفحة:
............
الجزء والصفحة:
............
 5-8-2018
5-8-2018
 5114
5114




+
-
20
Regioselective Enolate Formation and Diastereoselectivity
Lithium enolate derivatives of aldehydes or ketones may be formed at low temperature by slow addition of the carbonyl compound to an excess of LDA in THF (-78 ºC). In this procedure self aldolization is avoided, because freshly introduced aldehyde (or ketone) reacts with the powerful LDA base more rapidly than with any less basic enolate already present. In the following example, a preformed lithium enolate of 2-pentanone is reacted with propanal at low temperature, ca. -15 ºC. Only the desired aldol addition takes place, with very little enolate exchange occurring by proton transfer. Both enolate formation and aldol addition are essentially irreversible, with a forward to reverse rate difference of roughly ten powers of ten (based on pKa values). The lithium salt of the final adduct is quenched in dilute acid to give the β-hydroxyketone. A new stereogenic center is created in this reaction, resulting in a racemic mixture of products.
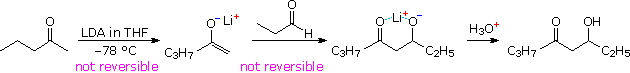
Aldol reactions at an α-methylene group generally create two new stereogenic centers, thus producing diastereomeric pairs of enantiomers. These diastereomers are called syn and anti, as described in an earlier section. The ability to control the configurational outcome of such reactions is vital to the use of aldol reactions in complex synthesis, and considerable effort has been directed toward achieving this end. The three reactions shown in the following diagram illustrate some of the factors to be considered. If the preformed lithium enolate of cyclohexanone reacts with benzaldehyde under rate controlling conditions (irreversible addition), the anti isomer is the preferential product (eq. 1). Under equilibrating conditions, the two diastereomers are produced in nearly equal quantity. Magnesium and zinc enolates may be prepared by reacting α-bromo carbonyl compounds with these metals, as illustrated in equation 2. The aldol addition in such cases usually requires a higher temperature. In all these examples the reactants are achiral and the products are racemic.
Moderately sized cyclic ketones can only generate one enolate stereoisomer (configuration E), but acyclic ketones may give E or Z-enolates. In the case of 2,2-dimethyl-3-hexanone, reaction with LDA gives the lithium Z-enolate (98%), as shown in equation 3. Here, the rate controlled aldol reaction strongly favors the syn-diastereomer, but under equilibrium control the anti-isomer is the dominant product. The size of ketone substituents also influences the isomer distribution of aldol products, as illustrated by the E-enolate reactions of the two compounds in equation 4.
From these examples it is clear that kinetic selectivity in the aldol reaction depends in large part on the configuration of the enolate reactant. The first of the following tables illustrates the dramatic influence of the accompanying carbonyl substituent (R) on the Z to E ratio of enolate species formed by reaction of a CH3CH2C=O moiety with LDA. The second table demonstrates how the amide base used for enolate formation may direct reaction exclusively to an E or Z-product, provided the other carbonyl substituent does not exert a controlling influence (e.g. tert-butyl). Identification of enolate species is usually achieved by trapping them as their silyl enol ethers.
| R = | C2H5 | (CH3)2CH | (CH3)3C | C6H5 | CH3O | (CH3)2N |
|---|---|---|---|---|---|---|
| Z:E Ratio | 23:77 | 60:40 | 99:1 | 98:2 | 5:95 | 97:3 |
| Base | LDA | LTMP | LTMP + LiBr | LHMDS | (Et3Si)2NLi | [Ph(CH3)2Si]2NLi |
|---|---|---|---|---|---|---|
| Z:E Ratio | 23:77 | 15:85 | 1:99 | 66:34 | 99:1 | 100:0 |
| LDA = lithium diisopropylamide LTMP = lithium 2,2,6,6-tetramethylpiperidide LHMDS = lithium hexamethyldisilazide | ||||||
An explanation for these interesting and useful characteristics requires consideration of the transition state for base induced enolate formation. Two putative transition states are displayed at the top of the following diagram. An early state that resembles reactants is on the left, while a late state resembling products is on the right. The product from each transition state is the enolate drawn in the green shaded box between them. Dialkylamide bases such as LDA and LTMP are powerful bases that react rapidly with simple aldehydes and ketones, presumably by an early transition state. The chief steric interactions (orange circles) are between R & R1 in the substrate, and between R2 & L (a substituent on the nitrogen base). The former is particularly strong in tert-butyl ketones, resulting in predominate Z-enolate formation (the oxygen group is smaller than tert-butyl). For less sterically demanding R groups, the R2:L interaction acts to favor the E-enolate, and may dominate when L is large (i.e. LTMP). This tendency is enhanced by added LiBr.
In contrast, hexaalkyldisilazide bases such as LHMDS, (Et3Si)2NLi, and [Ph(CH3)2Si]2NLi are much weaker bases (at least a million times) and will probably react by a later transition state. Here the Si–N bonds are longer than the C–N bonds of the amide bases, and the entire L2NH moiety is shifted away from the enolate substrate. Consequently, the L:R2 interaction is relatively unimportant, leading to a preference for Z-enolates.
Extensive study of aldol reactions with aldehyde acceptors has led to the general rule: Z-enolates favor syn-products and E-enolates favor anti products. To explain this stereoselectivity a chair-like transition state, called the Zimmerman-Traxler model, has been proposed, and examples are drawn beneath the enolization transition states in the above diagram. The lower energy transition state for the Z-enolate aldol has a gauche relationship between the phenyl and R2 groups, and this leads to the syn-product. Rotating the aldehyde 180º about the C=O creates an equivalent transition state for the anti-isomer, which suffers from a destabilizing 1,3-diaxial hinderance between the phenyl and R1 groups. The favored transition state for an E-enolate aldol has a similar gauche interaction, but leads to the anti-product. Once the new C–C bond has formed, the aldol product can relax into a chair-like chelate structure (drawn in the brown box on the right). In the case of the anti-product, all substituents are equatorial. The equivalent structure for the syn-isomer has an axial R2 group. In general, anti-aldols are more stable than their syn analogs. A link to a model for the chair-like transition state is provided below.
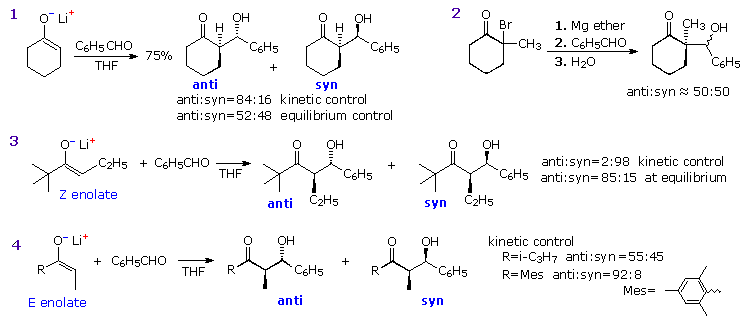
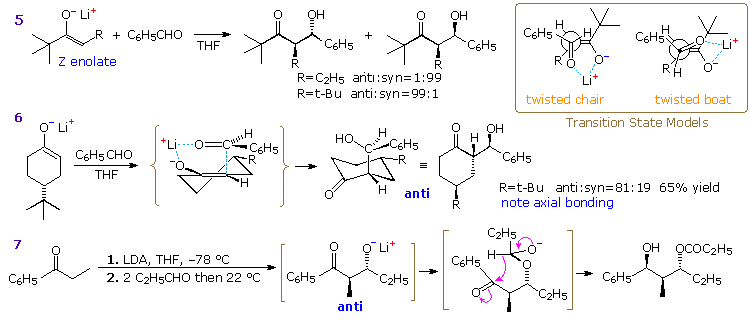
Three additional aldol reactions will be displayed above by clicking on the diagram. Reaction 5 demonstrates that a very bulky substituent on the α-carbon of a Z-enolate can dramatically change the stereoselectivity from syn to anti. A similar effect has been reported for E-enolate reactions. Two modifications of the idealized chair transition state, the twisted chair and boat models drawn in the box at the upper right, have been suggested to accommodate these facts. Note that different faces of the carbonyl group bond to the enolate species in these structures, with the re(si)-face in the twisted boat bonding to the re(si)-face of the Z-enolate to yield the anti-diastereomer.
Reaction 6 demonstrates stereoelectronic control in the bonding of the carbonyl reactant to the enolate intermediate. The bulky t-butyl substituent maintains an equatorial orientation on the six-membered ring, disclosing axial attack of benzaldehyde at the α-carbon. Finally, reaction 7 shows an intramolecular Tischenko reaction following an aldol reaction, the result being stereoselective construction of a 2-methyl-1,3-diol. The hydride transfer step is rate determining, so the aldol intermediate is the thermodynamically favored anti-diastereomer. A six-membered chair-like transition state accounts for the selective reduction of the carbonyl group.
A cautionary point must be made regarding these models and mechanisms. The lithium cation associated with the amide base and the enolates is organized in oligomeric clusters of substrate and solvent species that are much larger than the atoms (or groups) shown here. These amide and enolate ion clusters have variable compositions, and are presumably in rapid equilibrium with other clusters of the same type.
 الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















