























النبات

مواضيع عامة في علم النبات
الجذور - السيقان - الأوراق
النباتات الوعائية واللاوعائية
البذور (مغطاة البذور - عاريات البذور)
الطحالب
النباتات الطبية
الحيوان
مواضيع عامة في علم الحيوان
علم التشريح
التنوع الإحيائي
البايلوجيا الخلوية
الأحياء المجهرية
البكتيريا
الفطريات
الطفيليات
الفايروسات
علم الأمراض
الاورام
الامراض الوراثية
الامراض المناعية
الامراض المدارية
اضطرابات الدورة الدموية
مواضيع عامة في علم الامراض
الحشرات
التقانة الإحيائية
مواضيع عامة في التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحيوية والميكروبات
الفعاليات الحيوية
وراثة الاحياء المجهرية
تصنيف الاحياء المجهرية
الاحياء المجهرية في الطبيعة
أيض الاجهاد
التقنية الحيوية والبيئة
التقنية الحيوية والطب
التقنية الحيوية والزراعة
التقنية الحيوية والصناعة
التقنية الحيوية والطاقة
البحار والطحالب الصغيرة
عزل البروتين
هندسة الجينات
التقنية الحياتية النانوية
مفاهيم التقنية الحيوية النانوية
التراكيب النانوية والمجاهر المستخدمة في رؤيتها
تصنيع وتخليق المواد النانوية
تطبيقات التقنية النانوية والحيوية النانوية
الرقائق والمتحسسات الحيوية
المصفوفات المجهرية وحاسوب الدنا
اللقاحات
البيئة والتلوث
علم الأجنة
اعضاء التكاثر وتشكل الاعراس
الاخصاب
التشطر
العصيبة وتشكل الجسيدات
تشكل اللواحق الجنينية
تكون المعيدة وظهور الطبقات الجنينية
مقدمة لعلم الاجنة
الأحياء الجزيئي
مواضيع عامة في الاحياء الجزيئي
علم وظائف الأعضاء
الغدد
مواضيع عامة في الغدد
الغدد الصم و هرموناتها
الجسم تحت السريري
الغدة النخامية
الغدة الكظرية
الغدة التناسلية
الغدة الدرقية والجار الدرقية
الغدة البنكرياسية
الغدة الصنوبرية
مواضيع عامة في علم وظائف الاعضاء
الخلية الحيوانية
الجهاز العصبي
أعضاء الحس
الجهاز العضلي
السوائل الجسمية
الجهاز الدوري والليمف
الجهاز التنفسي
الجهاز الهضمي
الجهاز البولي
المضادات الميكروبية
مواضيع عامة في المضادات الميكروبية
مضادات البكتيريا
مضادات الفطريات
مضادات الطفيليات
مضادات الفايروسات
علم الخلية
الوراثة
الأحياء العامة
المناعة
التحليلات المرضية
الكيمياء الحيوية
مواضيع متنوعة أخرى
الانزيمات

Fluorescence Spectroscopy
 المؤلف:
F. X. Schmid
المؤلف:
F. X. Schmid
 المصدر:
In Protein Structure: A Practical Approach
المصدر:
In Protein Structure: A Practical Approach
 الجزء والصفحة:
الجزء والصفحة:
 11-5-2016
11-5-2016
 3433
3433




+
-
20
Fluorescence Spectroscopy
1. Fluorescence
Fluorescence emission of light is observed when, after excitation by the absorption of a photon, an electron returns from the first excited state back to the ground state. Absorption and emission are both virtually instantaneous processes and occur in about 10–15s. The average lifetime of the excited state is about 10–8s, which is a short time, but seven orders of magnitude longer than the time required for light absorption and emission. In the excited state, some energy is always lost by nonradiative processes (such as transitions between vibrational states). Therefore, the energy of the emitted light is always less than that of the absorbed light, and the fluorescence of a chromophore thus occurs always at greater wavelengths than its absorption. Fluorescence emission is much more sensitive to changes in the environment of the chromophore than light absorption. As the lifetime of the excited state is long, a broad range of interactions or perturbations can influence this state and thereby the emission spectrum. Fluorescence is thus an excellent and extremely sensitive probe to investigate the structure and function of proteins.
2. Fluorescence of Proteins
The aromatic amino acid residues phenylalanine (Phe), tyrosine (Tyr), and tryptophan (Trp) exhibit fluorescence emission when excited in the wavelength range of their absorption spectra. In proteins that contain all three aromatic amino acids, the emitted fluorescence is usually dominated by the contribution of the Trp residues, because both their absorbance at the wavelength of excitation and their quantum yield of emission are considerably greater than the corresponding values for Tyr and Phe. This is expressed by the “sensitivity” parameter (Table 1), which is 730 for Trp and 200 for Tyr. Phenylalanine fluorescence is not observed in native proteins, because its sensitivity of 4 is very low. In addition, Phe and Tyr fluorescence is also decreased by energy transfer to Trp residues.
Table 1. Absorbance and Fluorescence Properties of the Aromatic Amino Acids a
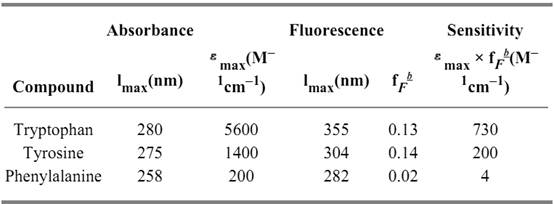
a In water at neutral pH; data are from (2).
b fF, fluorescence quantum yield.
Changes in protein conformation, such as unfolding, very often lead to large changes in the wavelength and intensity of fluorescence emission. The fluorescence emission of Trp residues in native proteins can be either greater or smaller than the emission of tryptophan in aqueous solution. Consequently, the fluorescence intensity can either increase or decrease upon protein unfolding. The emission maximum is usually shifted from shorter wavelengths to about 350 nm, which corresponds to the fluorescence maximum of tryptophan in aqueous solution. The exact location of this maximum depends to some extent on the nature and concentration of the buffer. In a hydrophobic environment, such as in the interior of a folded protein, tryptophan emission occurs at shorter wavelengths (indole shows an emission maximum of 320 nm in hexane). As an example, the emission spectra of native and of unfolded ribonuclease T 1 are shown in Figure 1. RNase T1 contains 9 Tyr residues and only one Trp (Trp59), which is inaccessible to solvent in the native protein.

Figure 1. Fluorescence emission spectra of native (—) and of unfolded (– – –) RNase T1. Native RNase T1 (1.4 µM( was in 0.1 M sodium acetate pH 5.0; the sample of unfolded protein contained 6.0 M GdmCl in addition. Fluorescence was excited at (a) 278 nm and ( b) 295 nm. The bandwidths were 3 nm for excitation and 5 nm for emission. Spectra were recorded at 25°C in 1 × 1 cm cells in a Hitachi F-4010 fluorimeter.
The fluorescence of the Trp residues of a protein can be investigated selectively by excitation at wavelengths greater than 295 nm. Because of the red shift and increased intensity of the absorbance spectrum of tryptophan when compared with tyrosine protein absorbance above 295 nm originates almost exclusively from Trp residues. A comparison of the emission observed after excitation at 280 and 295 nm gives information about the contribution of the Trp and Tyr residues to the observed fluorescence spectra. The data for RNase T1 in Figure 1 show that the shapes of the fluorescence spectra observed after excitation at 278 and 295 nm are virtually identical. The measured emission originates almost completely from the single Trp59, which is inaccessible to solvent and hence displays a strongly blue-shifted emission maximum near 320 nm. Tyrosine emission is barely detectable in the spectrum of the native protein, because energy transfer to Trp residues occurs. Unfolding of RNase T1 by guanidinium chloride results in a strong decrease in tryptophan fluorescence and a concomitant red shift of the maximum to about 350 nm. The distances between the Tyr residues and Trp59 increase upon unfolding, so energy transfer becomes less efficient. As a consequence, the Tyr fluorescence near 303 nm becomes visible in the spectrum of the unfolded protein when excited at 278 nm (Fig. 1a), but not when excited at 295 nm (Fig. 1b). The examples in Figure 1 indicate that, unlike absorbance, the changes in fluorescence upon folding can be very large, and the contribution of the Trp residues can be studied selectively by changing the excitation wavelength. Thus, fluorescence measurements are extremely useful to monitor conformational changes in proteins.
Multiple bands in an emission spectrum do not necessarily originate from different Trp residues of a folded protein. Figure 1 shows that even single Trp residues, such as Trp59 of RNase T1, can give rise to several emission bands. It indicates that the protein can adopt different conformations that equilibrate more slowly than the lifetime of the excited state (about 10–8s).
The fluorescence emission maximum of tyrosine remains around 303 nm, irrespective of its molecular environment. Therefore, the unfolding of proteins that contain no Trp residues is usually accompanied by changes in the intensity, but not in the wavelength of emission. As an example, the fluorescence emission spectra of the Trp59Tyr variant of RNase T1 (which is devoid of Trp residues( in the native and unfolded states are shown in Figure 2. A decreased Tyr fluorescence in the native state (as in Fig. 2) is frequently observed. It is thought to originate in the folded state from hydrogen bonding of the tyrosyl hydroxyl group and/or the proximity of quenchers, such as disulfide bonds.
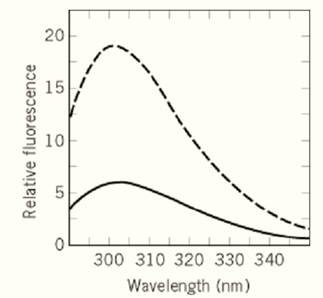
Figure 2. Fluorescence emission spectra of 1.5 µM of native (—) and of unfolded (– – –) Trp59Tyr-RNase T1. This protein contains 10 Tyr residues, but no Trp residue. The native protein was in 0.1 M sodium acetate, pH 5.0; the unfolded sample contained 6.0 M GdmCl in addition. Fluorescence was excited at 278 nm, the spectra were recorded as in Figure 1.
2.1. Environmental Effects on Tyrosine and Tryptophan Emission
The fluorescence intensity generally decreases with increasing temperature. This decrease is substantial, and to a first approximation, Tyr and Trp emission decrease by more than 1% per degree increase in temperature. Denaturants such as urea and guanidinium chloride also influence the fluorescence of Tyr and Trp (1).
3. Practical considerations
3.1. Fluorescence Spectrophotometers
Fluorescence spectrophotometers (“fluorimeters”) are single-beam instruments. The light from an intense source (usually a Xenon lamp) passes first the excitation monochromator to select the excitation wavelength, and is then focused into the center of the cuvette. The fluorescence emission is usually monitored at a right angle relative to the exciting beam; it passes through the emission monochromator and is detected by the sample photomultiplier. Most spectrofluorimeters operate in a split-beam mode, where a small portion of the incident light is directed to a reference photomultiplier to correct the fluorescence signal for inherent instabilities of the light source. To avoid baseline drift, some instruments interrupt the light beam periodically or employ pulsed lamps. The dark periods are used to adjust the baseline during the measurement. This increases the long-term constancy of the measured fluorescence.
3.2. Cuvettes for Fluorescence Measurements
Cuvettes for measuring fluorescence have polished surfaces on all four sides. Quartz cells are required for work in the UV region. For routine work, rectangular cuvettes (0.4 × 1 cm) are convenient. Since the exciting beam illuminates only the central part of the cell, the observed fluorescence intensity does not decrease necessarily when cuvettes with reduced cross sections are used. Cuvettes and glassware are easily contaminated with fluorescing substances that may leak out of plastic containers or are present in laboratory detergents. Before measurements, the cleanliness of the cell (and the distilled water) should be checked routinely by filling the cuvette with water and recording a blank in the wavelength range of interest. There should be no emission except the Raman peak of water.
3.3. Raman Peak of Water
In water, a Raman scattering peak is observed that is separated from the incident radiation by a fixed energy difference. After excitation at 280 nm (as in fluorescence experiments with proteins) the Raman peak of water is at 315 nm, ie, it overlaps with the emission spectrum of proteins. The Raman peak must therefore be subtracted from the measured fluorescence spectra. The performance of a fluorescence spectrometer can be checked easily by recording the signal-to-noise ratio at the Raman peak of water.
References
1. F. X. Schmid (1997) In Protein Structure: A Practical Approach, 2nd ed. (T. E. Creighton, ed.), IRL Press, Oxford, UK, pp. 261–297.
2. M. R. Eftink (1991) In Methods of Biochemical Analysis (C. H. Suelter, ed.), Vol. 35, Wiley, New York, pp. 127–205.
 الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
الاكثر قراءة في مواضيع عامة في الاحياء الجزيئي
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















