























علم الكيمياء

تاريخ الكيمياء والعلماء المشاهير
التحاضير والتجارب الكيميائية
المخاطر والوقاية في الكيمياء
اخرى
مقالات متنوعة في علم الكيمياء
كيمياء عامة
الكيمياء التحليلية
مواضيع عامة في الكيمياء التحليلية
التحليل النوعي والكمي
التحليل الآلي (الطيفي)
طرق الفصل والتنقية
الكيمياء الحياتية
مواضيع عامة في الكيمياء الحياتية
الكاربوهيدرات
الاحماض الامينية والبروتينات
الانزيمات
الدهون
الاحماض النووية
الفيتامينات والمرافقات الانزيمية
الهرمونات
الكيمياء العضوية
مواضيع عامة في الكيمياء العضوية
الهايدروكاربونات
المركبات الوسطية وميكانيكيات التفاعلات العضوية
التشخيص العضوي
تجارب وتفاعلات في الكيمياء العضوية
الكيمياء الفيزيائية
مواضيع عامة في الكيمياء الفيزيائية
الكيمياء الحرارية
حركية التفاعلات الكيميائية
الكيمياء الكهربائية
الكيمياء اللاعضوية
مواضيع عامة في الكيمياء اللاعضوية
الجدول الدوري وخواص العناصر
نظريات التآصر الكيميائي
كيمياء العناصر الانتقالية ومركباتها المعقدة
مواضيع اخرى في الكيمياء
كيمياء النانو
الكيمياء السريرية
الكيمياء الطبية والدوائية
كيمياء الاغذية والنواتج الطبيعية
الكيمياء الجنائية
الكيمياء الصناعية
البترو كيمياويات
الكيمياء الخضراء
كيمياء البيئة
كيمياء البوليمرات
مواضيع عامة في الكيمياء الصناعية
الكيمياء التناسقية
الكيمياء الاشعاعية والنووية

Photochemical Reactions: Alkene Isomerization
 المؤلف:
William Reusch
المؤلف:
William Reusch
 المصدر:
Virtual Textbook of Organic Chemistry
المصدر:
Virtual Textbook of Organic Chemistry
 الجزء والصفحة:
............
الجزء والصفحة:
............
 2-9-2018
2-9-2018
 16747
16747




+
-
20
Photochemical Reactions: Alkene Isomerization
A photochemical reaction occurs when internal conversion and relaxation of an excited state leads to a ground state isomer of the initial substrate molecule, or when an excited state undergoes an intermolecular addition to another reactant molecule in the ground state. The cis-trans photochemical isomerization of stilbene is a reaction of the first kind, as shown in the following diagram. Both cis and trans-stilbene undergo π → π* electron excitation by absorption of uv light. Whereas isolated double bonds require 180 nm light for such excitation, conjugation with the phenyl substituents lowers the transition energy to about 300 nm, a more easily achieved source. The molar absorptivity of the cis-isomer is less than that of the trans-isomer because steric crowding of the ortho sites causes the phenyl groups to twist slightly out of coplanarity.
The stability of the stereoisomers of stilbene is due to a 62 kcal/mole barrier to rotation about the double bond produced by the π-bond. This bonding is absent in the π → π* excited state (magenta curve in the diagram). Both the initial S1 states formed from the cis and trans ground states are slightly twisted (the cis by 25º & the trans by 13º) with the C=C double bond being lengthened by about 4.5%. These local S1 states quickly relax to a common lower energy twisted configuration (θ ≅ 90º). Non-radiative internal conversion of this S1 twisted state leads to the transition state region of S0, which decays equally to the ground states of the cis and trans isomers. This simple configurational isomerization about a single double bond is referred to as a One-Bond-Flip (OBF) event.
A small proportion (6%) of the trans-S1 state fluoresces back to the trans-isomer, but there is less than 0.1% fluorescence from the cis-S1 state. Triplet states (not shown) may also be formed, but there is no observable phosphorescence in solution, and non-radiative decay from such states results in similar cis-trans isomerization. The quantum yield of trans to cis isomerization decreases in viscous solvents, accompanied by an increase in fluorescent decay. Freezing the solutions to a rigid glass halts the isomerization and results in maximum fluorescence efficiency.
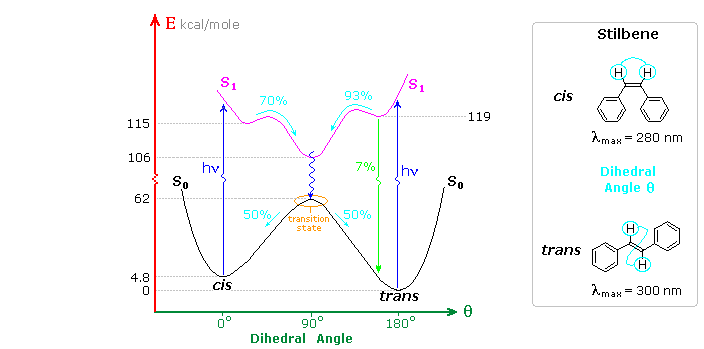
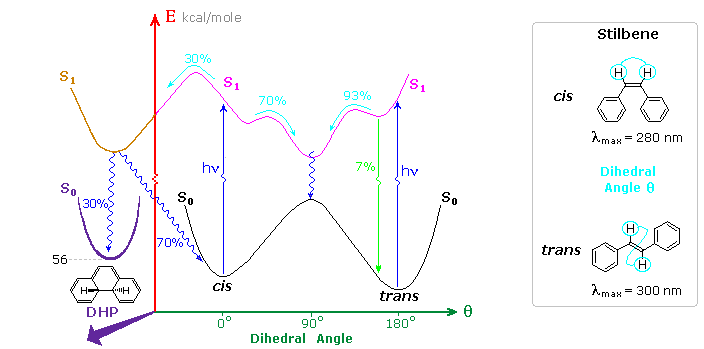
Inspection of the cis-S1 local minimum near θ = 0º shows that only 70% of the molecules in this state relax to the lower energy twisted S1 state. The remaining 30% undergo an electrocyclic rearrangement to an isomeric 4a,4b-dihydrophenanthrene (DHP) S1 state, as illustrated above.
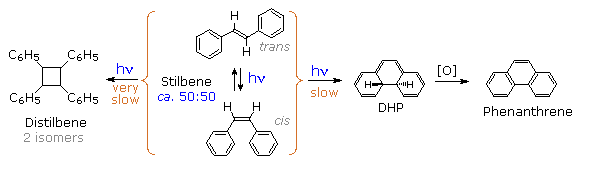
Molecules occupying this new excited state then relax to either DHP or cis-stilbene ground states. Over time, DHP accumulates and may be converted to phenanthrene by mild oxidation with air or iodine. Substituted phenanthrenes thus become available from trans-stilbene precursors prepared by Wittig or Grignard procedures.
Another minor product from the photolysis of stilbene has been identified as 1,2,3,4-tetraphenylcyclobutane (distilbene), shown in the following diagram. Unlike the previously described unimolecular isomerizations, this compound is formed by a bimolecular reaction between an electronically excited stilbene and a ground state stilbene molecule. The dimer accumulates slowly, irradiation of a 0.07 M solution of trans-stilbene in benzene producing 27% (as two stereoisomers) after two months of exposure. As expected, higher stilbene concentrations increase the rate of dimer formation. Thus, a 0.56 M solution of trans-stilbene in ethyl acetate yielded 11% dimer in four hours of irradiation. Mercury lamps were used as the uv light source in all these cases.
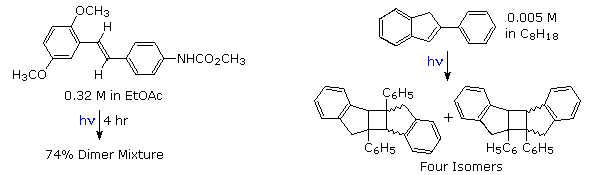
Electron donating substituents on the benzene rings facilitate the dimerization, as does restricting the ability of the double bond to isomerize or achieve orthogonally twisted excited states. Examples of these influences will be displayed above.
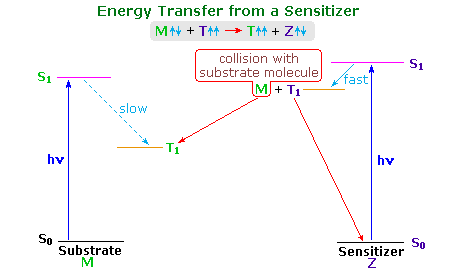
The stilbene reactions described above have been attributed to singlet excited states. Triplet excited states exist, but their formation by intersystem crossing is inefficient. In order to study the behavior of triplet excited states it is often necessary to generate them by energy transfer from a higher triplet excited state of a suitable sensitizer molecule. Spin conservation requires that spin exchange take place during the collisional energy transfer. Thus, a sensitizer triplet (Zt), generated from sensitizer molecule Zs, reacts with a ground state stilbene molecule (Ms) in the following manner:
Zt(↑↑) + Ms(↑↓) → Zs(↑↓) + Mt(↑↑).
The following diagram illustrates important features of this sensitization reaction. The ideal sensitizer (Z) absorbs light preferentially with respect to the substrate (M), and undergoes efficient intersystem crossing to a triplet excited state (T1). This energetic state then serves to activate a substrate molecule to a lower energy triplet state by collisional exothermic energy and spin exchange, returning the sensitizer to its ground state. A variety of useful sensitizers have been identified, and a few of these will be drawn below.
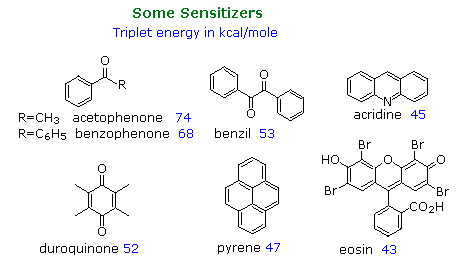
In the early 1960's Prof. George Hammond's group at California Institute of Technology undertook an extensive study of the photosensitized isomerization of stilbene and related alkenes and dienes.
|
Photosensitized Stilbene Isomerization |
||
|
Sensitizer |
Triplet Energy |
Cis:Trans Ratio |
|
benzophenone |
69 kcal/mole |
60:40 |
|
benzil |
53 kcal/mole |
85:15 |
|
pyrene |
47 kcal/mole |
90:10 |
|
eosin |
43 kcal/mole |
0:100 |
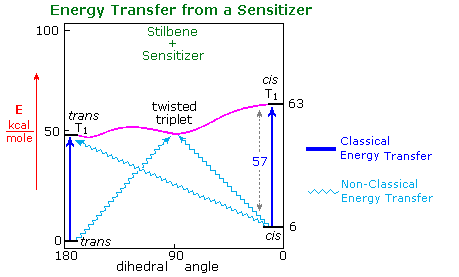
A few of their findings are presented in the table on the right, the same photostationary cis:trans ratio being observed from either isomer as the starting point. a diagram illustrating this system will be displayed above. The unexpected change in steady state isomer distribution with the triplet energy of the sensitizer could not be rationalized as a single classical energy transfer. All sensitizers having a greater triplet energy than 60 kcal/mole act by rapid collisional vertical energy transfer (heavy blue arrows) to the first encountered stilbene isomer. The initial cis and trans triplets undergo fast, and in the case of trans reversible, conversion to a lower energy twisted triplet, which then decays to cis and trans-stilbene at rates that favor cis by a factor of 1.5. As the triplet energy of the sensitizer drops below 57 kcal/mole, two changes occur. First, the efficiency of energy transfer to cis-stilbene diminishes and the trans isomer is selectively activated, resulting in a greater [cis]/[trans] photostationary state. Next, direct nonvertical excitation to the trans and twisted triplets may take place (light blue wavy arrows). These competing excitations and subsequent decay to cis and trans ground states lead to remarkable variations in isomer ratios. Finally, the very low energy triplet state of eosin is unable to effect any electronic excitation of stilbene, and simply catalyzes thermal equilibration, which strongly favors the trans-isomer (trans is 6 kcal/mole more stable than cis) These sensitized isomerizations do not give any significant amount of the electrocyclic rearrangement or dimerization reactions observed from singlet excited states.
 الاكثر قراءة في كيمياء عامة
الاكثر قراءة في كيمياء عامة
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















